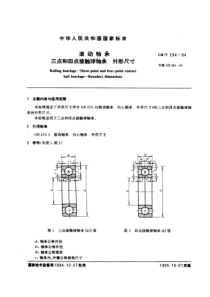
您好,欢迎访问三七文档
名词解释1、合理药物设计:根据药物发现过程中基础研究所揭示的药物作用靶点,即受体,再参考其内源性配体或天然药物的化学结构特征,根据配体理化性质寻找和设计合理的药物分子,以便有效发现、到达和选择性作用与靶点的又具有药理活性的先导物;或根据靶点3D结构直接设计活性配体。2、高通量筛选:HTS,以分子水平和细胞水平的实验方法为基础,以微板形式作为实验工具载体,以自动化操作系统执行实验过程,以灵敏快速的检验仪器采集实验数据,以计算机分析处理实验数据,在同一时间检测数以万计的样品并以得到的相应数据库支持运转的技术体系。3、药物的体内过程即A、D、M、E的中文名称及各自定义:分别为吸收:药物从用药部位进入体循环的过程。分布:药物在血液、组织及器官间的可逆转运过程。代谢:药物在吸收过程或进入体循环后,在体内酶系统、体液的PH或肠道菌从的作用下,发生结构转变的过程,此过程也称为生物转化。排泄:药物或其代谢物排除体外的过程。4、基于靶点的药物设计:TBBD,以生命科学为基础,根据疾病特异的功能、症状和机制,发现和研究药物作用靶点以及与预防相关的调控过程。5、基于性质的药物设计:PBBD,运用计算机辅助设计软件,根据配体的理化性质对设计的先导物结构预测它们的吸收、分布、代谢、排泄和毒性(ADME/T),估计药物在体内的释放度和生物利用度,判断类药性6、基于结构的药物设计:SBDD,以计算机辅助药物设计为手段,其方法分为基于靶点的直接药物设计和基于配体的简介药物设计两类,运用受体学说和分子识别原理,设计对受体进行调控的先导物,或根据已有药物作用力大小和构效关系判断来推测新化合物的药效,达到发现活性分子的目地。7、定量构效关系:QSAR,研究的是一组化合物的生物与其结构特征之间的相互关系,结构特征以理化参数、分子拓扑参数、量子化学指数和结构碎片指数表示,用数理统计的方法进行数据回归分析,并以数学模型表达和概括量变规律。8、三维定量构效关系:3D-QSAR,以配体和靶点的三维结构特征为基础,根据分子的内能变化和分子间相互作用的能量变化,将已知一系列药物的理化参数和三维结构参数与药效拟合出定量关系,再以此化合物预测新化合物的活性,进行结构的优化和改造。1、简述基于靶点结构的药物设计的基本流程。定义活性位点→产生配体分子→配体分子打分→合成及活性测定→先导物2、根据设计来源不同软药可以分为几种类型?软药和前药的区别有几个方面?软类似物;活化的软类似物;用控释内源物设计天然软药;活性代谢物;无活性代谢物等类型。区别:①先导物不一样,前药是以原药为先导物的,软药的先导物既可以是原药也可以是原药的代谢物;②作用方式不一样,前药在体外无活性,只有到达靶点释放出原药才有活性,而软药在体外是有活性的,它们到达靶点发挥治疗作用后一步代谢失活。3、简述先导物发现的可能途径。①筛选途径:从众多的化合物中运用生物筛选模型挑选有生物活性的先导物。现代筛选途径涉及组合化学、组合库和高通量高内含筛选。②合理药物设计:基于靶点和配体的作用机制、三维结构和识别过程以及与药物理化性质相关的体内过程,进行有的放矢的药物设计。4、药物作用的靶点的定义及理想的药物靶点特点是什么?靶点:也称靶标,指具有重要生理或病理功能,能够与药物相结合并产生药理作用的生物大分子及其特定的结构位点,这些生物大分子主要是蛋白质,有一些是核酸或其他物质。特点:①药物作用于靶点对疾病治疗的有效性②药物作用于靶点后引起的毒副反应小③便于筛选药物靶点的成药性。5、简述药效基团的虚拟筛选一般流程。小分子准备→产生构象→由活性分子生成药效基团的假设→优化、修改药效基团的假设→生成药效团模型→数据库搜寻(虚拟筛选)6、Lipinski的类药五倍律是什么?什么情况下该方法不适合预测药物的类药性?①相对分子质量<500(5×100);②氢键供体数<5个(5×1);③氢键受体数<10个(5×2);④lgP<5(若使用ClgP)或<4.15(若使用MlgP)(5×1)⑤分子内可旋转键数RB≤8或分子中环的数目rings≤4⑥化合物极性分子表面积PSA≤120A2当违反上述任意两个或多个规则时,化合物出现口服生物利用度差或代谢分布差的可能性就很大(大于百分之九十)对于抗生素、抗真菌药、维生素及强心苷等具有主动转运特征的药物不适用1、理想的药物靶点应具有哪些特点?(1)药物作用于靶点对疾病治疗的有效性。(2)中靶后引起的毒副作用反应小。(3)便于筛选药物的靶点成药性2、骨架迁越及在药物设计中的应用?骨架迁越:由苗头或先导化合物分子产生新结构的分子,保留原有的生物活性,通过结构骨架变换,连接适宜的药效团,产生新结构类型的药物,骨架迁越涉及丰富的药物化学内涵和技巧。应用:(1)将化合物转化成为类药分子-----改善药物动力学性质;刚-柔骨架的变换,改善药代性质;亲脂-极性骨架变换,改善溶解性和分配性;新的骨架若参与同受体结合,可改善与受体的亲和力;骨架适中的策略如果过小的骨架如苯环缺乏有用信息;过于复杂的骨架带来成本过高问题。(2)创制具有自主知识产权的新药或IP产品--破专利,Me-too,Me-better;3、前药设计应注意哪些原则?(1)在母体药物最适宜功能基处键合载体分子。(2)前药应无活性或活性较低,转运基团应无活性。(3)明确前药在体内的活化机制。(4)转化为母体药物的速度应该是快速动力学过程,并降低母体药物的直接代谢,以保证母体药物在靶点有足够的浓度。(5)应容易合成与纯化,最好是一步反应,且载体廉价易得。4、药物设计分三个不同的筛选途径是(基于功能途径、基于症状途径、基于机制途径)。5.举例说明软药设计原理设计原理为:(1)以某种药物的已知无活性的代谢物作为先导物。(2)将这种代谢物进行结构修饰,以获得无活性代谢物的结构类似物。(3)新的软类似物的结构设计应是经一步代谢就能产生原来无活性的代谢物,而不经过有毒中间体阶段。(4)对活化阶段的分子进行修饰,控制所设计的软药的转运、结合的性质以及代谢速率和药动学行为。泼尼松龙等在体内可发生17位的羟乙酮基氧化成无活性羧基,它是设计软药的良好先导物。将泼尼松龙酸与氯代甲醇成酯得软药氯替泼诺,则成为较少副作用的软药,用作滴眼剂,治疗炎性和过敏性眼疾患。氯替泼诺吸收入血液循环后,迅速代谢成预期无活性的代谢物,自尿和胆汁排出。治疗指数与氢化可的松比提高20倍。6.举例说明生物电子等原理。生物电子等排(bio-isosterism):当分子或基团的外层电子相似、或电子密度有相似的分布,分子的大小或形状相似,均可认为是生物电子等排体,它们的理化性质可能有较大的差异,但对同一受体发生相互作用,产生大致相似或相关的生物效应。双氢麦司昔摩和硫代麦司昔摩都是γ-氨基丁酸的环状类似物。环中的C=N部分是GABA分子中C=O的生物电子等排体,环中的S是环中O的等排体。两者在GABA-A受体上都有很强的激动作用。神经递质γ-氨基丁酸具有降低血压,安神,促进脑部血流,增进脑活力,健肝利肾,改善更年期综合症等作用.和胆汁排出。治疗指数与氢化可的松比提高20倍。7.设计“MeToo药物”主要有哪些策略和方法?现在所说的metoo药物,通常是指基于已经发现的药物的结构骨架,通过取代基团或侧链的变化,使得活性、生物利用度等提高或毒性降低,并绕开专利的药物。本质上是一类化学创新药,但与全新结构的新化学实体(newchemicalentity,NCE)相比,创新性略低。MeToo药物的研究设计中,使用的主要技术手段多为生物电子等排原理。一般可以从以下几个方面进行MeToo药物的设计。1.经常进行知识的追踪与更新,关注新出现的突破性新药,运用生物电子等排等理论,对新出现的化合物进行结构修饰和改造,希望以此能找到作用机制形同、相似甚至相反的新化学实体,用于疾病的治疗。2.在追踪新药的过程中,如发现尚无专利保护的NCE,要尽快进行结构改造和修饰,若能找到活性更优的化合物,要尽快申请专利,形成自己的知识产权保护。3.对已有专利保护的新药,要对专利进行深入调研,研究专利保护的范围,在不侵犯别人专利的情况下进行专利边缘的创新。我们可以从以下几个方面进行专利边缘的创新:(1)有意识的对化合物的局部化学结构进行改造,以改变药物的脂水分配系数、酸碱性、体内代谢转化的方向及延长作用时间等;(2)引入杂原子,甚至是稀有元素,以改变化合物的元素组成;(3)对同一领域的多个专利同时进行研究和调研,分析总结构效关系,通过拼合原理,运用药物化学和化学合成知识,将两部分或两部分以上的活性构象融合在一个新的化合物中;(4)重视手性药物的开发与研究,可以吧以前外消旋体供药的化合物进行拆分或进行手性立体合成,将异构体分开来研究,有可能会得到新的“MeToo”药物。总之,MeToo药物的设计,要求我们队专利进行深入的调研,撞我基本的专利法知识,运用药物化学、药理学、药物分析等药学知识,对专利已保护的化合物进行充分的研究,以突破专利的保护,设计出结构新颖的MeToo药物。酶抑制剂酶抑制剂:限制酶催化底物的反应能力作用基础:通过限制酶催化底物的反应能力,使底物浓度增高或代谢产物浓度降低,达到改善症状的目的酶抑制剂具备三种特征:1、结构上与底物或者中间产物相似;2、必须能够到达靶酶,并维持一定浓度;3、具有特异性,作用仅限于靶酶分类:1、可逆性抑制剂:竞争性抑制剂、非竞争性抑制剂、反竞争性抑制剂竞争性抑制剂:化学结构与底物相似,与底物竞争酶的活性部位可逆性抑制剂例子:抗痛风新药---非布索坦(非嘌呤类黄嘌呤氧化酶选择性强抑制剂);磺胺类药物(二氢叶酸合成酶抑制剂),作用机制:磺胺类药物能与细菌生长所必需的对氨基苯甲酸(PABA)产生竞争性拮抗,干扰了细菌的酶系统对PABA利用;抗艾滋病毒:齐多夫定(核苷类逆转录酶抑制剂,作用机制:抑制病毒复制过程所需的逆转录酶-阻碍病毒DNA的合成。2、不可逆抑制剂:与酶上的一个功能基形成共价键,使酶的结构和功能发生不可逆的变化而完全失活,即使加入大量底物也不能解除这种失活状态。根据不可逆抑制剂的作用机理的不同,基本上可分为3类:定向活性部位抑制剂、基于机理的抑制剂、伪不可逆抑制剂。基于机理的抑制剂具有以下三个方面的结构特征和性能:一是同正常底物的化学结构相似;二是在通常状态下,它们具有的低反应性能的潜在基团或中间体在没得催化阶段,经靶酶诱导激活,转化为反应性能强的活性基团或中间体;三是与酶的活性部位发生化学反应共价结合,使酶不可逆失活。例:β—內酰酶抑制剂(氧青霉烷类——克拉维酸、青霉烷砜类——舒巴坦)肽拟似物天然活性肽的缺陷:1、不稳定,易被酶水解;2、选择性差肽拟似物:生物活性肽经结构修饰获得的化合物称为肽拟似物。其比天然生物活性肽本身具有许多优点,如口服生物利用度高、耐受酶解、作用时间长、稳定性好、制成口服制剂病人易于接受,易贮存、无免疫原性,研究及生产成本低等。设计原理及方法:生物活性肽的环化——构象限制;限制氨基酸的取代;肽类二级结构的分子拟似物;酰胺键拟似物——假肽设计;类肽设计;CADDCADD:(computeraideddrugdesign)计算机辅助药物设计,以计算机为工具,采用各种理论计算方法和分子图形模拟技术,根据积累的大量有关结构和功能的资料,设计具有一定药效的新分子。CADD的主要目的是利用统计学和分子模型化技术来指导新的先导结构的设计或发现2组合化学的概念是什么?其合成新化合物总数的决定因素有哪些?答:组合化学是将一些基本分子构建模块通过化学或生物合成的手段,将他们系统的装配成不同的组合,由此得到大量具有结构多样性特征的分子,从而建立化学分子库的方法。其合成新化合物的总数取决于两个因素(1)每一步反应中应用化合物的种数(2)反应的步数3根据化合物库的来源不同,可以将发现先导化合物的方法分为哪些?答:(1)大范围多品种的随机筛选发现先导化合物(2)通过主题库的筛选发现先导化合物(3)基于已有知识进行定向筛选发现先导化合物4基于片段分子设计的研究方法是什么,它有什么优点?答:研究方法:(
 三七文档所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
三七文档所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
 扫描二维码
扫描二维码








 gt跑车浪漫旅
gt跑车浪漫旅
本文标题:药物设计学复习资料
链接地址:https://www.777doc.com/doc-2018815 .html