您好,欢迎访问三七文档
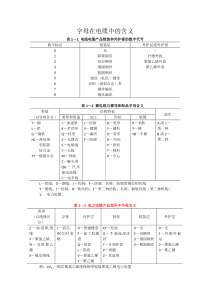
ACPDDRT-PCR技术摘要:随着基因组计划的顺利实施,大量的生物信息被解析,分离和鉴定差异表达基因已成为分子生物学研究的热点。mRNA差别显示技术(DDRT-PCR)是目前有效筛选、分离差异表达基因的方法之一。就DDRT-PCR的基本原理简要概述,阐明了该技术在生物生长发育、杂种优势、抗逆性基因研究中的应用等。关键词::mRNA差异显示技术杂种优势抗性ACP技术Abstract:WiththedevelopmentofGenomeProjectandanalysisofmassivebiologicalinformation,separationandidentificationofdifferenceexpressinggenehavebecomeahotissueinmolecularbiologyresearch1ThemRNAdifferentialdisplay(DDRT-PCR)isoneofeffectivemethodsforscreeningandisolatingdifferentiallyexpressedgenes.TheprinciplesofDDRT-PCRtechniquewereintroduced1.Theusabilityandachievedresearchresultsbyusingthismethodweresummarizedfromtheaspectsofricedevelopmentgene,heterosisgeneandtheresistancegene1TheprospectiveapplicationofDDRT-PCRinpaddyricemutantandresistancetoagriculturalchemicalsresearchwasalsoexploredinthepaper.Keywords:mRNAdifferentialdisplayHeterosisResistanceACP许多年以来,分离差异表达基因的方法仅限于差异筛选cDNA文库,直到1992年Liang等发明了一种检测基因转录模式的方法,即mRNA差异显示技术(mRNADifferentialDisplayReverseTranscriptionPCR,DDRT-PCR)[1],它是将mRNA反转录技术与PCR技术二者相互结合发展起来的一种RNA指纹图谱技术,目前已广泛应用于分离鉴定组织特异性表达的基因,差别基因表达(differentialgeneexpress)是细胞分化的基础[2]。mRNA差别显示技术正是对组织特异性表达的基因进行分离的一种快速而行之有效的方法。它第一次实现了同时快速灵敏地检测到真核细胞中大部分的差异表达转录体的目的[3]。近些年来,在分离和克隆差异表达基因的功能基因组学研究中,国内外学者又发展了多种分析方法,如,代表性差异分析、RNA指纹技术、RC4D、SAGE、抑制消减杂交技术、cDNA-AFLP、iAFLP及基因芯片技术等方法克隆差异表达基因[4]。这些方法的发明与应用,取得了许多成就,克隆了一系列的差异表达基因。这些方法各具独特优点,但同时也具有其自身不同的缺陷。这些技术的原理和优缺点比较已有许多综述总结。[5]引物退火时能否与其靶序列特异结合,是PCR能否成功扩增的关键因素之一,因此引物结构的优化非常重要。退火温度的高低决定引物是否能与其互补链完全结合,还是有一个或多个碱基的错配,因此通过调整退火温度就可以增加引物与模板结合的特异性[6]。许多研究者提出了各种办法来提高引物退火的特异性,如在引物的5’端增加一段通用引物序列,或加一段同聚物,或加上一段环状序列,以增加PCR扩增的特异性和退火的稳定性,但这些方法并不能消除初始反应的非特异性退火[7]。作者将介绍一种在DDRT-PCR基础上发展起来的对差异表达基因克隆的新方法,即引物退火控制技术(AnneallingControlPrimer,ACP)。该方法则可较好地消除初始反应的非特异性[8]。1ACP技术的基本原理ACP技术通过一种特殊引物,使得特异性和灵敏度同时兼顾。具体的引物结构是(图1):退火控制引物由独特的三部分组成,3'端和5'端部分由中间的调节部分连接。3'端部分是核心部分,是一段基因特异性结合段,能与模板完全互补,但长度较短(10bp)左右,这个长度的核酸退火温度基本在37℃左右,也就是普通随机引物的退火温度;中间调节的部分为多聚脱氧次黄苷[poly(dI)],一般由5个脱氧次黄苷(dI)组成,在控制引物的退火温度时起着关键的作用[9]。ACP技术主要由以下三个部分组成:逆转录合成第一条链和两个不同阶段的PCR反应;5'端部分是通用引物序列。这三部分的具体作用为:3'端的核心序列用于第一、第二轮扩增时,保证扩增的灵敏度;中间的调节部分是ACP技术实现的关键区域,作用主要有以下两方面:①提高核心序列识别模板时的退火温度,这是由于dI可以识别ATGC任意碱基,这样就将核心序列的退火温度提高到50℃左右,②连续的dI在较低温度时,形成一个泡状结构,而使通用引物序列卷曲起来,不参与退火识别,而在较高温度时形成线性分子,而使整条引物打开,参与退火识别;5'端的通用调节序列是高温识别的通用引物区。2ACP技术的优点2.1假阳性低假阳性高是目前克隆差异表达基因的瓶颈问题之一,ACP技术可使引物在初始反应与模板链特异结合,扩增出真实可靠的产物,从而降低了PCR结果的假阳性。[10]2.2不需要聚丙烯酰胺凝胶电泳只需通过琼脂糖凝胶电泳就足以进行分析ACP技术显著提高了PCR扩增的特异性和敏感性,使得PCR产物的特异性大为增强。通过普通脂糖凝胶电泳可以检测PCR产物,且脂糖凝胶电泳显示的条带可以直接用于Northern杂交分析。[11]2.3不需要特别的技术ACP是基于PCR和普通琼脂糖电泳的一种技术,简单容易操作。2.4重复性高由于ACP采用简单容易的操作就可得到结果,避免了繁复步骤引入误差的机会,使得试验结果的可重复性大大提高。[12]2.5速度快、经济实惠ACP技术在较短时间内就可克隆到可信的差异表达基因,且不需要花大量的时间来排除假阳性。2.6PCR产物分布范围广产物条带分布可为150bp~2kb,这样不仅增加了鉴定差异表达基因的机会,也为预测基因的功能提供了更多重要的序列信息,足以满足分析所需。[13]3ACPDDRT-PCR技术的应用ACP技术在差异表达基因克隆中的应用,目前报道的主要集中于哺乳动物(小鼠、牛和猪等)生殖发育相关的基因的研究[14],通过该技术克隆了许多差异表达基因,为深入了解哺乳动物早期生殖发育相关的分子调控机制提供了有利证据。4、操作步骤1.总RNA的提取(见Northern杂交)2.第一链合成:(1)总RNA样品2μg;cDNA合成引物1μl;加ddH2O补至5μl。将管标号为1A,2A,PCA等。PC为阳性对照。(2)混匀后稍离心。(3)70℃孵育3min,冰浴2min后稍离心。(4)准备dNTPmix(以5管为例)每管5管5×第一链缓冲液2μl10μldNTPmix(各5mM)2μl10μlMMLV逆转录酶(200u/μl)1μl5μl总5μl25μl(5)每管加入5μl,混匀后稍离心。(6)42℃孵育1h。(7)75℃10min终止反应后置冰上,稍离心。(8)将2μl反应液分别转至新管中(新管标号为1B,2B,PCB等)。(9)将78μlddH2O分别加入B管中,混匀。(10)将72μlddH2O分别加入A管中,混匀。(11)将所有cDNA稀释液-20℃保存待用。3.dd-PCR:以引物P1,T9为例说明(0.5mlPCR管,1代表对照组,2代表实验组)管号cDNA样品引物(1)1AP1,T9(2)1BP1,T9(3)2AP1,T9(4)2BP1,T9(5)H2OP1,T9(6)RNA1P1,T9(7)RNA2P1,T9以下为阳性对照反应体系(8)PC1AP10,T8(9)PC1BP10,T8(10)PC2AP10,T8(11)PC2BP10,T8(12)H2OP10,T8(13)PCRNA1P10,T8(14)PCRNA2P10,T8在PCR管中加入:①cDNA样品1μlP引物1μlT引物1μl②准备其它PCR试剂的混合液:(在另一管中加入)成分每管(μl)所需管数(n=14)10×buffer2.02nddH2O14.014ndNTPmix0.20.2nα-32PdATP0.40.4nTaq酶0.40.4n终体积17.017n混匀后稍离心。③将17μlPCR混合液加入各反应管,则各管总体积为20μl。④开始PCR扩增。⑤PCR循环参数:在GeneAmpPCRSystems2400&9600扩增仪上执行以下程序:94℃5min1cycles40℃5min68℃5min94℃30sec2cycles40℃30sec68℃5min94℃20sec23cycles40℃30sec68℃2min1cycles68℃7min。⑥反应结束后置-20℃保存备用。4.电泳和放射自显影(1)配制6%变性聚丙烯酰胺凝胶,灌注测序板。(2)预电泳30min。(3)每个dd-PCR反应取出5μl于一新微量离心管中,加5μlloadingbuffer,混匀,离心,置94℃变性2min。立即置冰浴。(4)停止预电泳,冲洗加样孔。(5)加样2μl。70W电泳2.75hr至二甲苯青到达胶的底部。(6)下胶:(同测序胶)(7)干胶:在胶表面覆上保鲜膜,80℃干胶30min。(8)X光片-70℃曝光过夜或更长时间(根据射线强度而定)。图2-6显示了dd-PCR放射自显影的X光片5.差异条带回收再扩增(1)X光片经D72显影、酸性定影液定影后,水洗晾干。置X光片灯上比较寻找差异条带。(2)用一次性手术刀片在胶上切割差异条带,加ddH2O20μl,沸水浴15min,离心取上清为模板。(3)以原引物扩增:回收DNA7μl10×PCRbuffer5μl5mMdNTPmix0.5μl引物P2.5μl引物T2.5μlTaq酶2μl加ddH2O至总体积50μl(4)执行PCR程序:93℃3min;93℃1min→60℃1min→68℃2min,20次循环;68℃5min。(5)取PCR产物10μl2%琼脂糖电泳检测。(6)PCR产物乙醇沉淀,10μlddH2O溶解。6.以PCR产物为探针,标记后进行Northern杂交,证实基因的真实性。7.PCR产物的TA克隆。8.DNA序列测定。9.检索分析。参考文献:1LiangP,PardeeA1Science,1992,257:967~97112StraussM,BauerD,MullerH1NucleicAcidsResearch,1993,21:4272~428013钱前,程式华,水稻遗传学和功能基因组学[M]1北京:科学出版社,200614HuWJ,ZhangSH,ZhaoZ,etal1PlantPhysiologyandMolecularBiology,2003,29(6):507~51415李竑,沈思师,许智宏,等1实验生物学报,2003,36(1):54~6016高风华,谭学林1云南农业大学学报,2006(2):150~15317EndoM,TsuchiyaT,WatanabeM,etal1GenesGenetSyst,2004,79(4):213~21618GuimilS,ChangHS,ZhuT,etal1PNAS,2005,102:8066~807019郭蕾,程英豪,王紫1北京大学学报(自然科学版),2006,42(2):175~79110林碧源1用DNA芯片鉴定水稻花器官发育相关基因[硕士学位论文]1福州:福建农林大学,2006111辛业芸,邓小林,罗崇善1杂交水稻,2007,22(6):1~5112贺滉,郭安平,孔华,等1安徽农业科学,2007,35(5):1307~1309113熊建华,陈学峰,张义平1中国水稻科学,2004,18(2
 三七文档所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
三七文档所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
 扫描二维码
扫描二维码





 qinmo21
qinmo21
本文标题:细胞与分子生物学
链接地址:https://www.777doc.com/doc-297259 .html