您好,欢迎访问三七文档
当前位置:首页 > 医学/心理学 > 基础医学 > 出血性疾病的诊断思路ppt-安徽省立医院
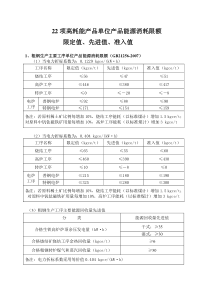
出血性疾病的诊断思路安徽省立医院血液科吴竞生MackmanN.BloodDisMolec36:104,2006MechanismsofthrombusformationFactorVII出血性疾病-基因突变/获得性因素引起凝血因子/血小板质或量异常.遗传性出血性疾病特点与危害性终身性致残性遗传性终生替代治疗家庭与社会沉重负担防治具有重要社会意义WorldHemophiliaDay2010,April17TheManyFacesofBleedingDisorders–UnitedtoAchieveTreatmentforAll”首次报告的罕见出血性疾病DisorderYearAuthorGlanzmannthrombasthenia1918GlanzmannFactorVdeficiency1947OwrenFactorVIIdeficiency1951AlexanderFactorXIdeficiency1953RosenthalCombinedFVandFVIIIdeficiency1954OeriFactorXdeficiency1956/7Telfer/HougieFactorXIIIdeficiency1960DuckertProthrombindeficiency1962(1947)Quick遗传性出血性疾病(安徽省立医院,2010,05)HA/HB910FXIDeficiency3FgDeficiency3FVIIDeficiency1FXIIIDeficiency2FXIIDeficiency2FVDeficiency2FXDeficiency1VWD40GT3AquiredHA15非血液科出血性疾病骨科-血友病(骨关节病变),最易误诊普外科-血友病,血管性血友病,FXIII缺乏(伤口不愈合),VK因子缺乏(肝胆疾病)。口腔科-血友病,FI缺乏,FV因子缺乏泌尿外科-血友病,FI缺乏,FVII缺乏。妇产科-VWD,血小板无力症一、出血性疾病的分类血管壁功能异常遗传性(毛细血管扩张症、家族、单纯性)获得性(过敏性紫癜、血管炎、免疫性)数量遗传性、获得性Aa、巨核、免疫性、ITP、DIC血小板异常数量(ET,CML)功能异常(GT、BS、颗粒性)凝血功能障碍遗传性(血友病、VWD、其他凝血因子缺乏)获得性(VK缺乏,肝脏病,DIC、药物)病理性抗凝物质凝血因子抑制物肝素样抗凝物质;狼疮样抗凝物纤溶亢进遗传性t-PA/u-PA↑抗纤溶酶获得(DIC,血栓症)(一)血管壁异常1、先天性或遗传性遗传性毛细血管扩张症;家族性单纯性紫癜;先天性结缔组织病2、获得性感染:如败血症;过敏:如过敏性紫癜;化学物质及药物:如药物性紫癜;营养不良:如VitC及PP缺乏症;代谢及内分泌障碍:如糖尿病,Cushing病;其他:如结缔组织病,动脉硬化,机械性紫癜,体位性紫癜等(二)血小板数量异常1、血小板数量异常(1)血小板减少①生成减少:如再生障碍性贫血、白血病、放化疗后骨髓抑制;②破坏过多:多与免疫有关,如原发性血小板减少性紫癜(ITP);③消耗过度:如弥散性血管内凝血(DIC);④分布异常:如脾功能亢进等(2)血小板增多原发性:原发性出血性血小板增多症继发性:如切脾后等血小板结构与功能血小板功能缺陷黏附异常巨大血小板综合征GP1b-FX血管性血友病(vWD)vWF量、质异常血小板型vWDGPIIb-FIX自发结合vWF聚集异常血小板无力症GPIIb-IIIa缺乏释放异常(颗粒异常)灰色血小板综合症粒储存异常或成分缺乏储存池病、致密体颗粒缺乏受体与信号传递异常环氧化酶缺乏症环氧化酶缺乏合成酶缺乏症合成酶缺乏信号传导缺陷症花生四烯酸不能动员CoagulationCascadeM-CPoon,MDSlide10(三)凝血异常1、先天性或遗传性(1)血友病A、B及遗传性FⅪ缺乏症(2)遗传性凝血酶原、FⅤ、Ⅶ、Ⅹ缺乏症,遗传性纤维蛋白原缺乏及减少症,遗传性FⅩIII缺乏及减少症2、获得性(1)肝病性凝血障碍(2)VitK缺乏症(3)尿毒症性凝血障碍等(四)抗凝及纤维蛋白溶解异常主要为获得性疾病肝素使用过量香豆素类药物过量及敌鼠钠中毒抗因子Ⅷ、Ⅸ抗体形成蛇咬伤、水蛭咬伤溶栓药物过量遗传性t-PA/u-PA↑抗纤溶酶(五)复合性止血机制异常先天性或遗传性血管性血友病(vWD)获得性弥散性血管内凝血(DIC)二.出血性疾病的诊断与分析病史+体征+实验室检测(一)病史出血史(出血年龄、部位)诱因(自发、创伤、手术)手术史(出血、伤口愈合)流产史(习惯性流产、死胎)药物史(阿司匹林、肝素、华发林、鼠药)家族史(出血史、近亲结婚)相关病史(肝、肾脏病)(二)临床表现出血部位:皮肤、黏膜、肌肉、关节、内脏特点:出血点、紫斑、血肿相关并发症:关节畸形、肌肉萎缩常见出血性疾病的临床鉴别血管性血小板凝血障碍性别女性多见女性多见多为男性家族史较少见罕见多见紫癜常见多见罕见大片瘀斑罕见多见可见血肿罕见可见常见关节罕见多见多见内脏偶见常见常见眼底罕见常见少见月经少见多见少见手术外伤少见可见多见1.表型诊断(筛选、定性、定量)2.分型(HA/HB)3.严重程度(轻、中、重)4.家系分析(检出携带者;高危胎儿产前诊断)5.基因诊断(直接、间接)(三)实验室检查建立完善的检测体系出血性疾病患者家系调查直接基因诊断临床表型诊断筛查凝血象血小板确定携带者核酸测序LD-PCR(1/22内含子倒位)遗传学控制确定突变基因早期治疗确诊凝血因子检测多重及长距离(LD-PCR)+核酸测序费时,昂贵,仪器FⅧ内含子1及22倒位技术比较快速,准确,易操作,无污染SouternBlot同位素,费时,复杂(三)实验室检查迅速、准确、实用、覆盖大部分出血原因1、筛选试验(1)血小板计数(2)出血时间(BT)(3)凝血酶原时间(PT))-组织因子途径(4)激活的部分凝血活酶时间(APTT)-内源凝血途径(5)凝血酶时间(TT)(6)纤维蛋白原(Fg)(2)出血时间(BT)指刺破皮肤自然出血到自然止血的时间。反映血管壁,BPC功能。传统BT已不能满足临床需要:Duck法:缺乏准确性,应淘汰。IVY法:敏感但难以标准化出血时间测定器法(1969):较敏感和准确第7届全国血栓与止血会议(关于出血时间和凝血时间的建议)BT不能废除,对评价血管及BPC功能有一定价值,对于预测手术过程出血并无帮助,建议不再作为术前常规,对如下疾病有诊断价值:VWD;血小板功能异常;低(无)FI血症;遗传性毛细血管扩张症;抗血小板药物等。血小板减少(假性vs真性)骨髓检查鉴别:生成破坏(免疫;非免疫性)PTAPTTPT、APTT、TT-NPLC-HMWKXIIPKXIIXVIIIVIIXVIIITT(3)凝血时间(CT)离体血液与异物表面接触,到血液自然凝固所需的时间,反映内源凝血系统整个凝血过程。玻片法:粗糙,敏感性差,仅测出FVIII0.15%,趋于淘汰。试管法:只能测出FVIII2%和FIX4%,建议保留,但不用于血友病诊断和肝素的检测。KPTT法:敏感,快速,建议代替其它CT。(1)血管壁异常:毛细血管镜、vWF、ET-1(2)血小板异常:形态、功能、PF3、PAIg、TXB2等(3)凝血异常:凝血活酶生成:FⅤ、Ⅶ、Ⅷ、Ⅸ、Ⅹ、Ⅺ、Ⅻ凝血酶生成:FⅡ、F1+2纤维蛋白原;Ⅰ(4)抗凝异常:AT、PC、FⅧ:C抗体(5)纤溶异常:3P、FDP、D-D、t-PA等(6)特殊检查2、确诊试验一期止血(毛细血管-血小板功能缺陷)结果判断与确诊试验PBC↓-骨髓生成障碍:AA,白血病,化疗后骨髓抑制消耗↑:DIC,TTP,HUS破坏↑:ITPPBC↑-骨髓增生性疾病(原发,继发)PBC计数正常+BT-功能障碍:聚集试验:ADP,胶原,AA诱导无反应-确诊血小板无力症GPIIb/IIIa无I血症I测定Ristocetin诱导无反应-VWDVWF测定BSSIb/X/VBPC正常+BT正常-血管性紫癜血小板计数出血时间测定减少正常外周血涂片血小板形态增大轻度增大/正常骨髓检查巨核细胞减少增多伴成熟障碍先天性无巨核ITP细胞性再障脾亢骨髓衰竭延长血小板聚集试验GTvWDvWF:Ag多聚体分析瑞氏托霉素辅因子试验二项聚集波缺如服用阿斯匹林血小板释放障碍无纤维蛋白血症初期止血障碍的实验诊断病例1、血小板无力症患者,男,17岁,自幼反复鼻出血、牙龈出血,黑便2天。父母近亲结婚,家族中无类似病史。查体:重度贫血貌,皮肤可见陈旧淤班,余正常。实验室检查:BPC84X109,血小板形态正常BT12’,PT12.3”,KPTT35”,vWF105%ADP诱导不聚集,Ristocetin诱导聚集正常,GPIIb/IIIa(CD41)13%,平均荧光强度1.32,诊断:GT,IDA治疗:输注血小板CoagulationCascadeM-CPoon,MDSlide10二期止血(凝血功能)结果判断与确诊试验APTT↑+PT(-):HA、HB、FXI缺乏、抗体,DIC、肝脏病(IX,XI)、抗凝剂、VWDFVIII、FIX、FXI、VWF、抗体测定PT↑+APTT(-):FVII、FV、FX缺乏、抗体,肝脏病、DIC、抗凝剂、溶栓治疗FVII、FV、FX、FVII、抗体测定APTT↑+PT↑:共同途径(FX、V、II、I):肝脏病、DIC、VK缺乏、内外凝血系统凝血因子、纤溶亢进PT+APTT均正常:FXIII缺乏部分凝血活酶时间(APTT)凝血酶原时间(PT)APTT延长PT延长APTT,PT延长FVIII,FIX,FXI测定FVIIIFIX正常HAHBFXI缺乏FVII测定FII,FV,FX纤维蛋白原缺乏凝血酶时间正常异常FII,FV,FX纤维蛋白原测定正常异常异常纤维蛋白原血症低纤维蛋白原血症先天性凝血异常的实验诊断PTTTAPTTPT-APTT,TT,PLC-NHMWKXIIPKXIIXVIIIVIIXVIIIFVII、FV、FX缺乏抗体,肝脏病DIC抗凝治疗PT纠正试验待测血浆+正常血浆吸附血浆诊断纠正不能纠正X不能纠正纠正V不能纠正部分纠正II病例2、遗传性FVII缺乏症男,23岁,血尿一个月于1991.11.20,入我院泌尿外科,请血液科会诊.追问病史,患者出牙,拔牙时多次发生牙龈渗血,劳累后关节肿痛。家族史:无类似症状患者。其父母为近亲结婚。查体:中度贫血貌,皮肤粘膜无出血点及紫癜。实验室:Hg90g/L,BPC100X109,尿WBC少许,RBC#,肾功能正常。PT↑,FVII:C0.1其母亲、二弟、二妹、表弟无出血,FVII为0.40;0.15;0.27;0.35,诊断:遗传性FVII缺乏症家系。治疗:PCC及血浆第二天血尿消失。APTT-PT,TT,PLC-N内源性凝血因子缺乏(HA、HB、FXI缺乏)抑制物(抗体)vWD抗凝剂(肝素)DIC、肝脏病PTTTAPTTHMWKXIIPKXIIXVIIIVIIXVIIIAPTT混合试验待测血浆+正常血浆吸附血浆诊断不能纠正纠正VIII纠正不能纠正IX纠正纠正XI、XII病例3、血友病A(轻型)患者,男,8岁,拔牙后牙龈出血12小时既往:无出血史,家族史阴性查体:无贫血,皮肤粘膜无出血点及瘀斑,关节无畸形。实验室检查:血小板、出血时间、PT、TT、Fg均正常APTT42”、vWF100%FVIII:C25%诊断:血友病A(轻型)治疗:DDAVP+止血芳酸APTT↑+无症状患者抑制物–狼疮抗凝物FXII缺乏轻度先天性内源性凝血因子缺乏病例4、遗传性FXII缺乏症患者,女,25岁,妊39周,拟行破腹产,术前凝血异常请血液科会诊。既往:无出血史。家族史阴性。父母近亲结婚。查体:无贫血,无出血。实验室检查:血小板、PT、TT、Fg均正常,APTT120”FVIII/FIX/FXI均正常FXII:C1%诊断:FXII缺乏症处理:无特殊,产后无出血病例5、原发性APS伴双侧耳廓坏死(中华耳鼻咽喉杂志2001,36(2):93)女,30岁.妊20周伴双侧耳廓和面
 三七文档所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
三七文档所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
 扫描二维码
扫描二维码








 563630645
563630645
本文标题:出血性疾病的诊断思路ppt-安徽省立医院
链接地址:https://www.777doc.com/doc-335756 .html