您好,欢迎访问三七文档
实验前准备根据自己实验的情况提前预定超净台进入细胞室之前必须穿鞋套,戴手套将实验所需要的实验仪器放入超净台内紫外照射灭菌15-20min高压灭菌操作1.将所需灭菌的枪头,EP管等仪器放入枪头盒或饭盒中,并用报纸将枪头盒包好。2.将仪器放入灭菌锅之前,首先要检测一下锅内的水位是否超过锅底的圆圈,若没有超过则需加水。3.检测水位之后,将包好的器材放入锅内,盖上锅盖并拧紧螺丝。4.插上电源,先将电压调至250V,目的在于排尽锅内的空气,(当排气阀有白气冒出时,说明空气已排尽)空气排尽后,关闭排气阀。5.关闭排气阀后指针上升,当指针竖直时,将电压调至125V,保持锅内压力的稳定。6.开始计时,灭菌30min。结束时关闭电源,等至指针慢慢降下即可。7.拿出锅内的器材放入恒温烘箱中烘干,2天后即可拿出。(组织内)蛋白质提取过程1.对癌组织和癌旁正常组织进行称重,记录数值。2.根据RIPA裂解液Ⅲ说明书(每100mg组织中加入1ml溶液A,1ul溶液B,5ul溶液C)按照组织块得大小配好ABC混合液。3.将组织块加入匀浆器中,并加入相应量得ABC混合液,冰上静置5-10min,开始研磨。4.将研磨好的组织液倒入事先准备好的EP管中,12000rpm/5min.5.离心结束后,收集上清液,弃去沉淀,上清即为所要的蛋白。蛋白质浓度测定1、取96孔板冲洗干净后超声处理30min,之后用去离子水冲洗干净烘干即可2、配制BCA标准蛋白液:浓度原液体积去离子水体积A管:2000ug/ul30ul0ulB管:1500ug/ul37.5ul12.5ulC管:1000ug/ul25ul25ulD管:750ug/ul18.75ul31.25ulE管:500ug/ul12.5ul37.5ulF管:125ug/ul3.125ul46.875ulG管:25ug/ul0.625ul49.375ulH管:00ul50ul取8个EP管,分别标上以上字母,并按上表加入相应的原液蛋白和去离子水先向96孔板内加入200ul的绿色底物溶液(根据实验计算所需要用孔的个数加入底物液)加完后,将配好的A,B,C,D,E,F,G,H标准液10ul按照96孔板上第1列上的字母标记依次加入之后在其他的孔内加入10ul蛋白样品(每个样品应重复2次)加好样品后取保鲜膜将96孔板封闭好,以防止蛋白浓度发生变化,之后将板放入保温箱中静置30min30min后将96孔板拿出放入测量仪器内(应按照仪器使用说明进行操作)数据测完后另建文件夹,保存数据,之后插入尤盘拷取数据在获得的数据结果中,根据BCA的标准蛋白浓度梯度值和560nm下对应的OD值计算可得出一个OD值与浓度相关的标准曲线:y=ax+b(计算机上操作)将测得的蛋白样品的OD值带入公式,即可求出蛋白样品的浓度若实验有实验组和对照组,应将两组的蛋白浓度标准化,即是两组的蛋白浓度相同(相同浓度下的两组实验才具可比性)将浓度标准化的样品蛋白与loadingbuffer混匀至100ul(混合液中loadingbuffer应为1X的,但实验室的一般为5X),稀释方法:取20ul5Xloadingbuffer+80ul样品蛋白混匀即可在加样做westernblot之前,蛋白样品需变性:将与loadingbuffer混匀好的蛋白样品放入100度金属浴内高温变性10min(EP管上要加帽以防止温度过高EP管盖会冲开)(组织内)RNA提取过程1.将组织放入匀浆器中,加入1ml的Trizol,冰上静置10min,进行研磨。2.将匀浆好的上清转移至1.5mlEP管中,进行离心12000rpm/5min.3.离心后收集上清液于新的EP管中,加入200ul氯仿,剧烈摇匀后静置5min,然后12000rpm/5min.4.离心后取上清于另一新的EP管中,取液时不要取到中间层的蛋白。之后按1:1的比例加入异丙醇,摇匀后静置15min,进行离心12000rpm/10min。5.离心后倒掉上清(可看到白色RNA沉淀),加入1ml75%乙醇DEPC水溶液温和震荡,悬浮沉淀,3000rpm/5min.6.倒掉上清,在滤纸上空干。然后加入20ulDEPC水溶解沉淀,并吹打均匀。(如果RNA量较多,可相应的加更多的DEPC水,40ul甚至60ul)7.测定RNA浓度,取1ulRNA+99ulDEPC水于EP管中(相当于稀释100倍),另取一EP管放100ul的DEPC水用于定标。定标后倒掉,用去离子水冲洗比色皿,然后测定RNAOD值和浓度,记下数据。8.RNA电泳(用于检测提取的RNA是否已降解)配胶(1%的胶):取0.2g琼脂糖+20mlTAE+1ulEB制胶时,应让液体沸腾3次,冷却后加入EB混匀(每20ml加1ulEB),胶制好后倒入板中凝固,并垂直地插入梳子,静置30min后拔下梳子(可提前用梳子划动下胶,使得胶更均匀)。取1ul的Loadingbuffer和样本RNA混匀,加样正确的电泳结果应有3条带:28S、18S、5S。28S亮度应为18S亮度的2倍,5S条带越宽越代表RNA降解越多。如果加样孔内有残留代表蛋白污染,如果离加样孔很近的地方有条带代表DNA污染。细胞冻存首先向培养瓶内加入胰酶(1ml)消化细胞显微镜下观察细胞形态,当细胞变圆脱落时,即可用适量培养基中止消化,之后将细胞液吸入15ml的管内(动作要快,防止胰酶损害细胞)将管内1/3的细胞液放回培养瓶继续培养(剩余的细胞液平均冻存两管)离心剩余的细胞液,1200rpm,5min(用等量的PBS配平)离心后弃上清,得白色沉淀。可在试管台上轻轻来回划动试管,使得沉淀松动更易于悬浮用配好的3ml冻存液悬浮沉淀,吹打混匀,使得细胞为单个细胞分别取1.5ml放入两冻存管内,在冻存管上做好标记:细胞类型,代数,日期等标记好后4°冰箱放置30min(切勿超过30min),计时之后-20°冰箱中放置30min最后放入-80°冰箱内可保存半年。若半年后还没有使用则需要将细胞拿出来复苏,长满后再冻存起来。冻存液的配制方法:血清(20%)+DMSO(10%,超净台上锡纸包裹的)+培养基(70%)例如配3ml冻存液:血清=600ul;DMSO=300ul;培养基=2100ul细胞复苏将实验所需要的仪器提前放入超净台内紫外灭菌15-20min在整个复苏过程中冻存的细胞都应保持在37°水浴中烧杯中放入37°水,将从冰箱内拿出的细胞用镊子夹住放入烧杯内摇晃使细胞慢慢融化细胞融化后,用酒精浸湿的纸擦干冻存管之后在超净台内将细胞液吸取到放有适量(例10ml)PBS的15ml管内吹打均匀(在取细胞之前提前在管内倒入PBS)1200rpm,5min离心,离心后弃上清得沉淀将得到的细胞沉淀用适量的培养基悬浮,并吹打均匀,使细胞单个存在吹打均匀后,用吸管将细胞液吸入培养瓶中(大/小培养瓶根据自己的实验而定)最后向培养瓶内加入适量的培养基显微镜下观察细胞情况最后将培养瓶盖子拧松,放入37°培养箱培养WesternBlottingWesternblot实验过程中所需要配制的试剂:1XTBST:(用于洗膜)100ml10XTBS+900ml去离子水+1mlTween205%牛奶:取5g牛奶溶于1000ml1XTBST中混匀1XRunning:(用于跑电泳)10XRunning100ml溶于900ml去离子水中1Xtransfer:(用于转膜)10XTransfer100ml+100ml无水甲醇+800ml去离子水一抗:用5%牛奶将一抗稀释2000倍(稀释倍数根据自己实验而定),若抗体由甘油保存,稀释一抗应取所计算体积的2倍。二抗:用5%牛奶将二抗稀释10000倍(牛奶封闭性更好)具体操作流程:1.清洗玻璃板一只手扣紧玻璃板,另一只手蘸点洗衣粉轻轻擦洗。两面都要洗净,之后先用自来水冲洗干净,再用去离子水冲洗干净,立在桌面上晾干。2.配胶(区别于RNA电泳胶)玻璃板对齐后放入夹中卡紧。然后垂直卡在架子上准备灌胶。先按照10%Gel(10ml)比例配下层的分离胶,加入TEMED后立即摇匀即可灌胶。灌胶时可用10ml枪吸取5ml胶沿玻璃缓慢放出,待胶面升到绿带中间线高度即可,然后加入100ml异丙醇封闭静置等待胶凝固,(若试管内剩余的胶凝固代表玻璃板内的胶已经凝固了)凝固后倒掉异丙醇,用去离子水冲洗干净,并用吸水纸将水吸干,并避免刮破胶再按照5ml的比例配上层的浓缩胶,加入TEMED后立即摇匀后即可灌胶。将配好的胶灌满剩余的空间,之后垂直缓慢地将梳子插入浓缩胶中,避免产生气泡胶凝固后即配胶结束配胶注意事项:若配好的胶不立即使用,可以保存在4度冰箱内,但保存时间不宜过长(用卫生纸包裹玻璃板,浸湿卫生纸,放入一次性塑料手套内即可,此时不要拔下梳子)蛋白胶分为两层,上层为浓缩胶,下层为分离胶。按照10%Gel的方法进行配胶。注意:选择玻璃板时要看清玻璃板上的大小标记,一般用10ml的。玻璃板一定要用去离子水洗干净,之后室温晾干。浓缩胶和分离胶配制所用Tris-Hcl的PH值是不同的,分别为7.8和6.83.跑电泳与上样配制(1000ml)1XRunning电泳缓冲液:(实验室现有的是10X)取100ml10XRunning缓冲液溶于900ml去离子水中即可得到1XRunning缓冲液将配好的胶板放在电泳槽内,注意正负电极的准确连接,两手分别捏住梳子的两边竖直向上轻轻将其拔出倒入配好的电泳缓冲液,若使用的是两块胶,加入的电泳液液面应超过“2Gel”字符线(电泳液也要灌满玻璃板)加蛋白上样量:测完蛋白含量后,计算含20-50ug蛋白(根据自己的实验需要进行选择,没有固定的量)的溶液体积即为上样量。用10ul枪头进行加样,将枪头插入加样孔缓慢加入样品,加样过快的话容易使样品冲出加样孔(注意同一实验和对照组所加的上样量应该相等,这样才具有可比性),同时加入2-3ulMarker作为条带分子量大小的对照加样完毕后,盖上电泳槽盖,连接电源,先调电压至120V,按start开始跑电泳当样品被压成同一水平的线时,可以降低电压至100V或80V左右,电泳至样品内的溴酚蓝刚跑出即可终止电泳,关闭电源,进行转膜4.转膜转膜前先用镊子捏住两张硝酸纤维素膜的一边浸泡在无水甲醇中配制1Xtransfer(转膜缓冲液):取10xtransfer100ml+100ml无水甲醇+100ml去离子水混匀即可将转膜液倒入搪瓷盘内,并放入转膜所需的夹子(夹子具有黑白两面)、两块海绵垫(海面上带有滤纸)、小绿板和浸过的膜将夹子打开使黑的一面位于底部保持水平,在其上垫一张海绵垫,用小绿板来回擀几次排除其内的气泡。要先将玻璃板撬掉才可剥胶,撬的时候动作要轻,除去小玻璃板后,将浓缩胶用小绿板轻轻刮取,要避免不要把分离胶刮破。小心剥下分离胶盖于滤纸上,用手调整使其与滤纸对齐,轻轻用小绿板擀去气泡。之后将另一海绵垫平铺在胶上,用手压住,并用小绿板赶走气泡,注意动作要轻柔,避免胶与膜发生错位。气泡赶净后就可以合起夹子(整个过程都要在转膜液中进行)。另外一块胶按照形同的过程进行操作之后将夹子放入转移槽中,要使黑色的面对槽的黑面,夹子的白面对槽的红面。由于在转膜的过程中会产生热量,所以将整个转膜槽放入装有冰块的桶内进行。插上电源,调电压至80V,转膜2h(转膜时电极方向注意是膜正胶负)转膜结束后,可看到膜上有明显的Marker条带,用丽春红染色液让膜显色,之后可以看到膜上蛋白的条带将膜放入一平皿内于摇床上脱色,放入1xTBST洗三次,每次10min。若事先知道目的蛋白的分子量,可以用保鲜膜将膜包裹,之后将膜修剪脱色之后,用牛奶对膜过夜封闭或封闭1h(4度冰箱内的摇床上过夜)封闭后,用TBST洗3次,每次10min加一抗2h(若牛奶封闭1h,可以加一抗过夜)用1xTBST洗3次,每次10min加二抗,封闭1h用1xTBST洗3次,每次10min5、
 三七文档所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
三七文档所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
 扫描二维码
扫描二维码



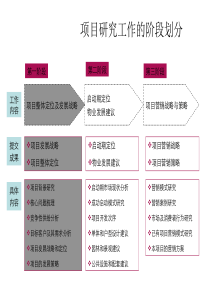



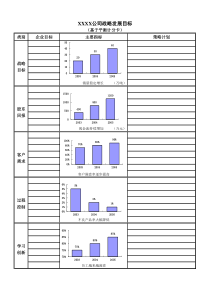

 panging
panging
本文标题:常见细胞实验流程
链接地址:https://www.777doc.com/doc-7105225 .html