您好,欢迎访问三七文档
当前位置:首页 > 行业资料 > 国内外标准规范 > 化学药物杂质研究的基本思路及案例分析 李眉
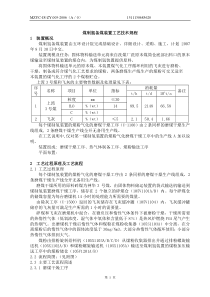
2011-04-261化学药物杂质研究的基本思路及案例分析李眉中国医学科学院医药生物技术研究所化学药物杂质研究的基本思路及案例分析李眉中国医学科学院医药生物技术研究所2011-04-262主要内容一.概述二.国内外对药物杂质研究的相关技术要求三.杂质研究的基本思路四.新法规下杂质研究评价的新动向2011-04-263一.概述1.杂质定义、分类及来源2.杂质研究的重要性3.目前杂质研究存在的主要问题2011-04-261.杂质定义、分类及来源杂质定义—任何影响药物纯度的物质ICH---药物中存在的,化学结构与该药物不一致的任何成分1)有毒副作用的物质2)本身无毒副作用,但影响药物的稳定性和疗效的物质3)本身无毒副作用,也不影响药物的稳定性和疗效,但影响药物的科学管理的物质2011-04-26按理化性质分类—有机杂质、无机杂质、残留溶剂有机杂质:包括工艺中引入的杂质和降解产物等,可能是已知的或未知的。化学结构与活性成分的分子式类似或具渊源关系,通常称为有关物质。无机杂质:原料药及制剂生产或传递过程中产生的杂质,通常是已知的。主要包括:反应试剂、配位体、催化剂、重金属、其它残留的金属、无机盐、助滤剂、活性炭等。残留溶剂:原料药及制剂生产过程中使用的有机溶剂,一般具有已知毒性。2011-04-266按照来源分类—工艺杂质、降解产物等工艺杂质:工艺过程中引入的杂质,包括反应物、中间体、副产物、试剂、催化剂等降解产物:由药物降解产生,如水解、氧化、开环、聚合等反应产物,与药物的结构特征密切相关2011-04-267按化学结构分类—几何异构体光学异构体聚合物其他甾体其他氨基酸2011-04-2682.杂质研究的重要性保证药品安全有效是药品研发及药品评价所要遵循的一个基本原则;药品质量的稳定可控是保证药品安全有效的前提和基础;杂质研究是药物质量研究的一项重要内容;杂质研究及控制---是药品质量保证的关键要素之一2011-04-269杂质研究是一项系统工程。与工艺研究、质量研究其他项目、稳定性研究、药理毒理及临床研究间存在着密切关系,直接关系到上市药品的质量及安全性。药理活性或毒性杂质—安全性普通杂质,控制纯度—有效性杂质产生的原因—优化工艺,提高生产水平2011-04-2610严重的“药害”问题---引发社会的高度关注:新鱼腥草素钠注射液亮菌甲素注射液注射用甲氨蝶呤2011-04-2611实例1.新鱼腥草素钠注射液七十年代通过审批,上市剂型变化:2ml/支,10ml/支,20ml/支50ml/瓶,100ml/瓶;据SFDA数据库统计:目前生产该品种的厂家:92家;2011-04-2612《药品不良反应信息通报》第四期(2003.8)截止2003年第一季度,国家药品不良反应监测中心数据库中有关新鱼腥草素钠注射液引起的不良反应病例报告共272例,以过敏反应和输液反应为主,其中严重不良反应有过敏性休克12例、呼吸困难40例。2011-04-2613(至06年5月31日)病例报告总数严重病例报告过敏性休克死亡静脉注射445521611331肌内注射35210例数用药途径严重病例剂量范围静脉注射:20-250ml肌内注射:2-10ml对病例报告进一步统计分析对病例报告进一步统计分析2011-04-2614原因分析文献调研:新鱼腥草素钠注射液成份复杂文献报道:用气相色谱-质谱联用方法鉴定本品有效成份,鉴定出48种化合物。主要有效成分是癸酰乙醛(鱼腥草素)和甲基正壬酮(2-十一烷酮)癸酰乙醛(鱼腥草素)易于分解,氧化、脱氢后可以转化为甲基正壬酮过敏原??(成份、杂质、降解产物?)2011-04-2615实例2.齐二药事件2006年4月底,广东中山三院传染病科先后出现多例急性肾功能衰竭,高度怀疑与齐齐哈尔第二制药有限公司生产的亮菌甲素有关。5月3日上午,国家ADR中心获知,检索国家中心数据库及相关文献--没有类似报道。2011-04-26165月5日,调查认为:此事件的发生与齐齐哈尔第二制药有限公司生产的亮菌甲素注射液(批号:06030501)相关,且不排除与原料药、辅料及加工或合成过程等环节的相关性;5月9日,广东省药检所在亮菌甲素注射液中检出二甘醇。5月11日,黑龙江省药检所检验结果:批号为06030501的亮菌甲素注射液2ml和10ml规格中,检出二甘醇。存在问题:原料药控制不严2011-04-2617实例3.甲氨蝶呤事件7月6日下午,收到广西省药品不良反应监测中心报告的广西医科大学第一附属医院鞘内注射甲氨蝶呤、阿糖胞苷、地塞米松、生理盐水后10名患儿出现软瘫的不良事件报告;随后收到广西中心报告的“关于甲氨蝶呤出现多起不良事件的紧急报告”;不久,又获悉上海市使用同企业、同批号产品也出现类似不良事件;2011-04-26188月31日,国家局和卫生部决定暂停上海医药(集团)有限公司华联制药厂生产的注射用甲氨蝶呤和注射用盐酸阿糖胞苷用于鞘内注射。9月5日,国家局和卫生部联合发出通知:暂停上海医药(集团)有限公司华联制药厂注射用甲氨蝶呤和注射用盐酸阿糖胞苷(均为冻干粉针剂)的生产、销售和使用。2011-04-26199月14日,国家局公布调查结果:甲氨蝶呤、阿糖胞苷鞘内注射后引起的损害,与两种药品的部分批号产品中混入了微量硫酸长春新碱有关。存在问题:一条生产线生产不同药品的混入现象3.目前有关物质研究存在的主要问题研究基础薄弱杂质来源不清杂质检查方法缺乏针对性杂质检查结果难以评价需要提高杂质研究水平,包括杂质检查方法和验证水平。存在的主要问题:问题之一:未进行有关物质(包含异构体检查、β-内酰胺抗生素聚合物)检查研究实例.布洛芬缓控胶囊参考CP,原料药采用TLC法,制剂未规定有关物质检查,而USP、BP原料药、片剂、口服混悬剂均采用HPLC法检查有关物质;相关文献显示:可能存在杂质4-异丁基苯乙酮具有中枢神经系统毒性.问题之二:未进行方法的比较、优选研究,所用方法不能够有效检出药品中存在的杂质实例.氟哌利多注射液光照10天,UV法测含量下降了10%,采用TLC法未检出杂质斑点;而参照USP标准采用的HPLC法检查时,注射剂中杂质含量高达7.6%.问题之三:未进行充分的验证研究,所提供的资料不能证明方法的可行性实例:注射用阿魏酸钠阿魏酸钠对光较敏感,其有关物质检查需要特别关注光降解产物峰的分离情况。一些厂家通过调整流动相比例,致使降解产物被主峰掩盖,但又没将建立的方法和中国药典2005年版质量标准中2种色谱条件进行详细的对比研究。且光照10天后,含量下降3.8%,有关物质仅增加0.5%;问题之四:系统适用性不符合要求,色谱系统、操作等存在问题,导致试验数据不可靠实例:氟马西尼注射液稳定性资料各时间点未检出有关物质,但主峰保留时间波动大,图谱记录时间短,进样量偏低,试验结果不可靠。后来发现是系统老化,温度变化较大的问题;问题之五:杂质水平高于上市产品,忽视杂质的安全性论证,杂质限度的确定缺乏充分依据实例:罗库溴铵收载于BP/EP:杂质A不得过0.2%,杂质B、C不得过0.3%,杂质D、E、F、G均不得过0.1;其他单一杂质不得过0.1%,总杂质不得过1.5%.有2家申报品种采用了和BP/EP类似的色谱条件,可是有关物质实际测定结果远远超出了BP/EP同品种的限度要求,但未提供相关资料证明杂质的安全性。质量标准中有关物质限度过宽,限度设定缺乏充分的依据。2011-04-2626二.国内外对药物杂质研究的相关技术要求2011-04-2627ICH:2002年2月及2003年2月颁布---《新原料药的杂质研究指导原则》(简称Q3A(R))《新制剂的杂质研究指导原则》(简称Q3B(R))2011-04-2628有机杂质:1)特定杂质(SpecifiedImpurities)指在质量标准中分别规定了明确的限度,并单独进行控制的杂质。包括:----结构已知的杂质----结构未知的杂质对于结构未知的杂质,为便于在标准中进行指认,一般采用代号(如未知杂质A等)或合适的定性分析指标(如相对保留时间为0.8的杂质)加以区分2011-04-26292)非特定杂质(UnspecifiedImpurities)指在质量标准中未单独列出,而仅采用一个通用的限度进行控制的一系列杂质,其在药品中出现的种类与几率并不固定由于非特定杂质的不确定性,因此,在药品的临床前与临床研究中,很难对这些杂质的安全性进行评价ICH的指导原则对其限度做了明确的规定----在原料药质量标准中任一单个非特定杂质的限度不得过鉴定限度----在制剂的质量标准中仅控制降解产物,在制剂质量标准中任一单个非特定降解产物的限度不得过鉴定限度2011-04-2630报告限度(ReportingThreshold):超出此限度的杂质均应在检测报告中报告,并应报告具体的检测数据。鉴定限度(IdentificationThreshold):超出此限度的杂质均应进行定性分析,确定其化学结构。质控限度(QualificationThreshold):质量标准中一般允许的杂质限度,如制订的限度高于此限度,则应有充分的依据。2011-04-2631最大日剂量报告限度鉴定限度质控限度≤2g0.05%0.10%或1.0mg(取最小值)0.15%或1.0mg(取最小值)2g0.03%0.05%0.05%原料药的杂质限度2011-04-2632制剂的杂质限度报告限度最大日剂量≤1g1g限度0.1%0.05%鉴定限度最大日剂量1mg1mg~10mg10mg~2g2g限度1.0%或5ug(取最小值)0.5%或20ug(取最小值)0.2%或2mg(取最小值)0.10%质控限度最大日剂量10mg10mg~100mg100mg~2g2g限度1.0%或50ug(取最小值)0.5%或200ug(取最小值)0.2%或3mg(取最小值)0.15%2011-04-2633FDA:仿制药申请:原料药杂质研究指导原则(讨论稿)2005年1月28日在FDA网站发布2011-04-2634杂质的合理控制(QUALIFICATIONSOFIMPURITIES)A.质控限度的考虑ICHQ3A(R)中推荐的“质控限度”是根据原料药每日剂量来制订的。如果所制订的限度超过该限度值,就必须提供所订限度的合理性依据。某些情况下,“质控限度”可调高或降低。比如,当有证据表明某药物中的杂质与副作用相关,就很有必要降低该杂质限度。相反,如果杂质与安全性无多大关联,杂质限度值可以设定高一些。FDA会根据患者人群、药物分类及历史数据等因素考虑申请者对杂质限度的调整。2011-04-2635B.杂质限度的研究方法如果杂质水平超过了ICHQ3A(R)中推荐的“质控限度”,那么可以参考附录1中的决策树来制订杂质的合理限度。杂质限度研究应考虑诸多因素,包括患者人群、每日剂量、给药途径和用药持续时间等。杂质限度研究可以用含有拟控制杂质的原料药,也可以直接用分离出来的杂质进行。杂质限度研究可以采用下面三种方法:2011-04-2636第一.对比分析法ANDA中原料药的杂质可以采用相同的分析方法(如HPLC研究方法),与FDA已批准的同品种进行对比研究。一般情况下,已批准同品种应选择参比产品(RLD)。如果无法获得RLD药物,也可以与具有相同给药途径的药物进行对比研究(如片剂与胶囊比较)。建议用具有可比性的样品(如样品的留样时间要一致)进行稳定性研究,以获得有意义的杂质对比研究结果。如果原料药杂质水平与FDA已批准的同品种的杂质水平相当,那么可以认为该杂质得到合理控制。2011-04-2637第二.科学文献和主要代谢物法如果已定性杂质的水平得到科学文献的充分论证,那么该杂质的限度就无需进一步论证。如果某杂质本身也是原料药在体内的主要代谢物,通常也认为该杂质已得到合理控制。第三.遗传毒性研究法考虑到遗传毒性试验既费时间又代价不菲,此法一般是在前两种方法都无法对杂质合理研究论证才
 三七文档所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
三七文档所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
 扫描二维码
扫描二维码








 hehe13212
hehe13212
本文标题:化学药物杂质研究的基本思路及案例分析 李眉
链接地址:https://www.777doc.com/doc-9180985 .html