您好,欢迎访问三七文档
当前位置:首页 > 医学/心理学 > 药学 > 蒽及醌类成分文献综述
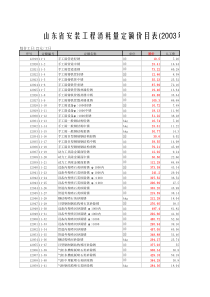
蒽及醌类成分分析摘要:综述蒽醌类化合物的测定方法的最新进展,包括荧光分析法、分光光度法、高效液相色谱法(HPLC)、毛细管电泳法(CE)、质谱联用法等,举例证明了每种分析方法的特点,为富含蒽醌类成分的药材和制剂的质量评价提供参考。关键词:蒽醌类荧光分析法分光光度法HPLCCE质谱联用法醌类化合物[1,2]是天然产物中一类比较重要的活性成分,是指分子内具有不饱和环二酮(醌式结构)或容易转变成这样结构的天然有机化合物。醌类化合物主要包括苯醌、萘醌、菲醌、蒽醌等类型;蒽醌类成分包括蒽醌衍生物及其不同程度的还原产物,例如氧化蒽酚、蒽酚、蒽酮以及蒽酮的双聚物等。蒽醌类化合物一般为黄色、橙色或红色,许多蒽醌类化合物具有生理活性。蒽醌类化合物大致分布在30余科的高等植物中,含量较多的有蓼科、鼠李科、茜草科、豆科、百合科、玄参科等,在地衣类和真菌中也有发现。各种蒽醌及其苷都具有多方面的生物活性,主要表现为降血脂、降胆固醇、利尿、抗氧化、抗过氧化等重要作用。但是蒽醌类化合物也有多种毒性作用,主要引起胃肠的各种不适,严重的可能会导致胃肠出血、呼吸困难、心悸和流体损耗。因此各种类型中成药、保健品中蒽醌类物质的质量控制十分重要。目前分析蒽醌类成分含量的方法有荧光分析法、分光光度法、高效液相色谱法(HPLC)、毛细管电泳法(CE)、质谱联用法等,本文就其进展进行概述。1、荧光分析法荧光分析法是材料元素分析的一种方法,它是利用一定波长的x射线照射材料,元素处于激发态,从而产激发出光子,形成一种荧光x射线。由于不同元素的激发态的能量大小不一样,所以产生的荧光x射线不同,进而根据荧光x射线的波长和强度,得出元素的种类和含量。物质的相对荧光光谱可作为定性和定量分析的重要手段,蒽醌类属于多环芳烃,有较强的荧光。荧光分析法的灵敏度高、选择性好、专属性强、信息量丰富、检测限低等优点,已被广泛用于药物的测定工作。物质的相对荧光光谱可作为定性和定量分析的重要手段,蒽醌类属于多环芳烃,有较强的荧光。相对于经典荧光法,同步荧光法在提高选择性和灵敏度等方面均有显著效果,更适合于混合物的直接分析。陈伟、林新华、黄丽英、罗红斌[3]等应用等吸收点双波长分光光度法对芦荟中芦荟苷含量测定进行研究。方法:依据芦荟苷与芦荟大黄素的光谱特征,361.4nm为测定波长,480.9nm为参比波长。结果:芦荟苷线性范围1×10-6~1×10-4mol/L,回收率为97.4%~101.6%,RSD为0.55%~0.74%,芦荟大黄素对测定不干扰结论:本方法简便可靠,重现性好,为芦荟资源的进一步开发利用及质量控制等提供新的方法和重要依据。2、分光光度法分光光度法是通过测定被测物质在特定波长处或一定波长范围内光的吸光度或发光强度,对该物质进行定性和定量分析的方法。在分光光度计中,将不同波长的光连续地照射到一定浓度的样品溶液时,便可得到与众不同波长相对应的吸收强度。用分光光度法测定蒽类成分的含量,常以1,8-二羟基蒽醌为参考标准,利用蒽醌类成分在碱性溶液中变红的反应,进行测定,此法的关键在于蒽醌类成分的提取及显色。Paris等认为中药大黄,以硫酸水解,氯仿提取较好,用乙醚提取则杂质较多。一般以5%氢氧化钠-2%氢氧化氨(1:1)的混合液为显色剂,因其稳定性差,不同时间吸收度不同,从而影响结果。陈军等[4]、马长清等[5]对分光光度法进行了改进,改进后的方法操作简便,结果可靠,稳定性好,不易受日光照射影响,弥补了碱液法显色后不稳定、易衰减的缺点。林新华等[6]运用双波长标准分光光度法同时测定芦荟中蒽醌类化合物芦荟大黄素甙和芦荟大黄素的含量,达到改善选择性又不降低灵敏度的目的,样品加标回收率均在100%±5%范围内。3、高效液相色谱法(HPLC)HPLC是近年来发展最快和使用最广的分析方法,具有分离效能高、分析速度快、选择性好和检测灵敏度高等优点,在有效成分定量分析中最为常用,其中又以反相高效液相色谱(RP-HPLC)法应用最广。钱启辉,田莉,陈象清[7]等人建立高效液相色谱法测定肠必清肠溶颗粒剂中番泻苷A、B含量。实验以C18柱为分析柱,以50%乙腈(含0.25mmol/L十六烷基三甲基溴化铵和0.25mmol/L磷酸二氢钠,每200ml加入冰醋酸75μl)为流动相,流速为1ml/min,检测波长为270nm。结果:在0.25μg/ml~80.0μg/ml浓度范围内呈线性关系(番泻苷A:r=0.9997;番泻苷B:r=0.9997),番泻苷A、番泻苷B测定方法重现性较好(RSD<2%)的紧密度均<2%,回收率均>98%。朱铁梁,张静泽,高文远,陈虹[8]等,采用高效液相色谱法(HPLC)测定大黄和大黄甘草汤样品中水解蒽醌和游离蒽醌的含量。使用的色谱柱为KromasilC18(250mm×4.6mm,5μm);流动相:A相甲醇;B相1%冰醋酸溶剂系统,线性梯度洗脱,流速1.0ml/min,检测波长为254nm,柱温35℃。结果:本文建立了HPLC方法检测并比较大黄和大黄甘草汤中结合型蒽醌以及游离型蒽醌的含量,精密度、重复性以及稳定性RSD均小于3%,加样回收率为97.39%~104.43%。研究结果表明大黄药材单煎或与甘草合煎样品中检测到芦荟大黄素,大黄酸,大黄素,大黄酚及大黄素甲醚5个共有峰,其含量分别为大黄中游离型蒽醌总量为(10.383±0.114)mg/g,结合型蒽醌总量为(7.315±0.082)mg/g;而大黄甘草汤中游离型蒽醌总量为(12.587±0.112)mg/g,结合型蒽醌总量为(8.299±0.054)mg/g,大黄单味药材相及与甘草配伍后样品中蒽醌类成分含量存在差异。大岛俊幸等[9]用反相高效液相色谱法对何首乌和夜交藤中的主要有效成分大黄素、大黄素甲醚、大黄酚及大黄酸等蒽醌成分进行定量分析,以TSK-gel80Ts为固定相,甲醇-水-磷酸(720:280:1)为流动相。样品用70%甲醇提取浓缩后用氯仿萃取,可分离出游离型和结合型蒽醌的定量用试液。以往,是根据总蒽醌含量和游离型蒽醌含量的差值求得结合型蒽醌的含量,而本文是在同一样品中直接测定结合型和游离型蒽醌的含量,此法也适于测定其它含蒽醌类成分的生药。4、毛细管电泳法(CE)4.1毛细管区带电泳法(CZE)毛细管区带电泳法(CZE)又称毛细管区带电泳自由溶液电泳,是毛细管电泳最基本,应用最广泛的一种分离模式。它的分离依据是基于试样组分质荷比的差异。JunkoKoyama等[10]利用环式糊精(CD)改造的毛细管区带电泳法对天然药物中大黄素、大黄酸及其糖苷进行同时测定,电解液由0.03mol/L四硼酸钠中加入适当体积的10%氢氧化钠、0.005mol/Lα-CD和10%乙腈组成。大黄素、大黄素-8-β-D-糖苷、大黄酚、大黄酚-8-β-D-糖苷的线性范围分别为5~50、25~150、5~75、50~500μg/ml,大黄素和大黄酚的加标回收率分别为96.1%~105.3%、99.2%~103.3%。4.2胶束电动毛细管色谱法(MEKC)MEKC是将离子型表面活性剂加到缓冲液中,如果浓度达到或超过临界胶束浓度,表面活性剂的单体就结合形成胶束,而溶质则在流动的水相和起固定相作用的胶束相之间分配,并由于其在胶束中不同的保留能力而产生差速迁移。大黄中的5种蒽醌类衍生物能在10min内,在含有25mmol/L脱氧胆酸钠的50mmol/L硼酸-氢氧化钠(pH=11)缓冲溶液中成功分离,但其中大黄酚和大黄素甲醚重现性不佳。此方法回收率高于90%,芦荟大黄素、大黄素、大黄酸的检测限分别为0.66、0.94、0.96μg/ml,线性范围分别为2.64~21.12、5.64~56.4、4.80~33.60μ/ml,加标回收率分别为90.61%(RSD=1.45%)、93.39%(RSD=1.21%)、92.31%(RSD=1.10%),日内和日间重现性RSD在0.96%~1.85%之间。此方法分析时间短,所需样品量少,抗污染及抗干扰能力较高,故分析中草药较合适[11]。刘训红,李俊松,张月婵,蔡宝昌,尹娣等[12],建立胶束电动毛细管色谱二极管阵列检测法(MEKC-DAD)同时测定大黄及其炮制品中大黄素、芦荟大黄素、大黄酸、大黄酚及大黄素甲醚含量。选择胶束电动毛细管电泳分离模式,用未涂渍标准熔融石英毛细管(75μ×64.5cm,有效长度56cm)为分离通道,以25mmol.L-1硼砂-25mmol.L-1SDS-10%乙腈(pH10.90)为背景电解质溶液,运行电压为12kV,运行温度为24℃,检测波长为254nm,压力进样为5kPa,5s。结果:大黄素、芦荟大黄素、大黄酸、大黄酚及大黄素甲醚浓度分别在2.4~47.0,2.4~48.4,2.1~41.4,1.9~37.0,3.8~75.0μg/ml范围内呈良好线性关系(r≥0.9994),加样回收率(n=5)分别为99.5%(RSD=3.7%),101.4%(RSD=2.4%),98.5%(RSD=3.8%),101.3%(RSD=3.9%),101.4%(RSD=3.1%)。说明该方法简单、准确,重现性好,可用于大黄饮片内在质量的评价和控制。4.3毛细管电色谱法(CEC)毛细管电色谱(capillaryelectrochromatography,CEC)以内含色谱固定相的毛细管为分离柱,兼具毛细管电泳及高效液相色谱的双重分离机理,既可分离带电物质也可分离中性物质。毛细管电色谱法是用电渗流或电渗流结合压力流来推动流动相的一种液相色谱法。因此,毛细管电色谱法可以说是HPLC和HPCE的有机结合,它不仅克服了HPLC中压力流本身流速不均匀引起的峰扩展,而且柱内无压降,使峰扩展只与溶质扩散系数有关,从而获得了接近于HPCE水平的高柱效,同时还具备了HPLC的选择性。颜流水,王宗花,罗国安,王义明等[13],在进一步改进仪器结构的基础上,将梯度加压毛细管电色谱用于同时分离大黄片提取液中5种蒽醌类活性成分,以建立适于中药等复杂体系中多组分分离分析的快速有效的新方法。实验结果显示,大黄提取液中的5种蒽醌化合物可在22min内完全分离,梯度洗脱微柱液相色谱的柱效为等度洗脱微柱液相色谱的6.63倍,梯度毛细管电色谱的柱效为梯度微柱液相色谱的4.6倍。5、质谱联用法近年来,高效液相色谱-质谱(HPLC-MS)联用,尤其是高效液相色谱-质谱-质谱(HPLC-MS-MS)[14]联用,已经成为发展最快的分析技术之一[15]。HPLC-MS[16]主要由HPLC仪、接口(HPLC与MS之间的连接装置)、质量分析器、真空系统和计算机数据处理系统组成。混合样品通过液相色谱系统进样,由色谱柱分离。从色谱仪流出的被分离组分依次通过接口进入MS仪的离子源处并被离子化,然后离子被聚焦于质量分析器中,根据质荷比而分离,分离后的离子信号被转变为电信号,传送至计算机数据处理系统,根据MS峰的强度和位置对样品的成分和结构进行分析。目前常用的HPLC-MS联用仪具有两大分类系统,一种是从MS的离子源角度来划分,包括电喷雾离子(ESI)、大气压化学电离(APCI)和基质辅助激光解吸离子化(MALDI)等;另一种是从MS的质量分析器角度来划分,包括四级杆质谱仪(Q-MS)、离子阱质谱仪(IT-MS)、飞行时间质谱仪(TOF-MS)、傅立叶变换质谱仪(FT-MS)。余勤.向瑾[17]等人建立了用HPLC-MS/MS法测定大鼠血浆中大承气汤蒽醌类组分浓度的方法。以布洛芬为内标,样品经盐酸酸化后,用乙酸乙酯萃取。以YMC-PackODS-AC18色谱柱(5µm,150×4.6mm)为分析柱,流动相为甲醇5mmol/L,乙酸铵(92:8,v/v,pH为5.6),流速为0.4ml/min.大承气汤蒽醌类组分的日内RSD小于5.69%,日间RSD小于6.23%,方法回收率在96.26%~108.95%,萃取回收率在59.36%~84.42%。质谱法(MS)、氢谱法(HNMR)、碳谱法(CNMR)和核磁法(DEPT)联用可对蒽醌类物质进行定性分析。结
 三七文档所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
三七文档所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
 扫描二维码
扫描二维码








 muygang
muygang
本文标题:蒽及醌类成分文献综述
链接地址:https://www.777doc.com/doc-2024504 .html