您好,欢迎访问三七文档
当前位置:首页 > 医学/心理学 > 药学 > 我国建立药品审评和审批质量管理规范的必要性和内涵
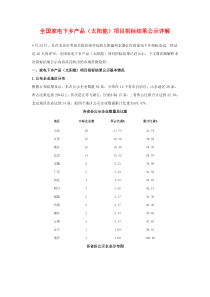
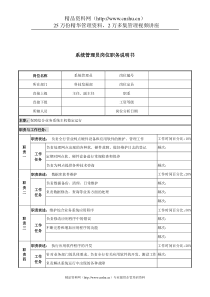
摘要在全球范围内,为了最大程度地保护和促进公众健康,制药工业都是一个受各国政府高度监管的行业。包括我国在内的世界各国都对药品的上市准入采取许可制度。绝大部分国家需要对企业所提交的上市申请进行独立的技术审评和审批。虽然国际人用药品注册技术要求国际协调会(ICH)的成立和推广极大程度上地统一了各国对药品在不同地区/国家申请批准时所需的数据要求,但在全球范围内,无论是工业界还是药品监管机构都意识到,监管机构在对上市申请的审评质量、审评效率、审评结果的明确性/可预见性和透明度方面都有待提高。近年来,美国食品药品监督管理局(FDA)率先试图通过建立“药品审评质量管理规范(GRP)”,以应对这样的挑战,并取得了良好的成效。本论文通过对美国FDA已建立的GRP及配套法规,以及这些法规实施后产生的影响的介绍,结合本企业在我国药品申报和注册中观察到/遇到的一些现象/案例、国家药品监督管理局(SFDA)和药品审评中心(CDE)公开发布的一些数据,试图对我国药品监管机构(SFDA及其下属的CDE)在审评质量、审评管理规范性方面存在的问题和挑战进行诊断和分析。结合我国国情以及行业特点,参照国外药品监管机构已制订的“药品审评质量管理规范”以及细则,展望在我国建立“药品审评和审批质量管理规范(GRRP)”的必要性和内涵,并提出具体的框架。旨在为我国的药品审评/审批建设规范的审评程序、提高审评质量,效率,可预见性和透明度见言献策。同时,“药品审评和审批质量管理规范”在我国的建立也将为工业界的药品研发提供明确的指南,促使我国的新药研发尽早与发达国家接轨,实现真正地自主创新。关键词:审评;监管机构;规范;质量;-TheNecessityandImplicationtoEstablish“GoodRegistrationandReviewPractice”inChinaAbstractTheNecessityandImplicationtoEstablish“GoodRegistrationandReviewPractice”inChinaTobestprotectandpromotepublichealth,pharmaceuticalindustryishighlyregulatedbyrespectivegovernmentworldwide.IncludingChina,pharmaceuticalproductshouldbeapprovedbyrespectiveregulatoryagencybeforeitsmarketentry.Regulatoryagenciesofmostcountriesperformindependenttechnicalreviewandapprovaltodataincludedintomarketingapplicationofapharmaceuticalproductsubmittedbyapplicant.AlthoughICH’sestablishmentandspreadgreatlyharmonizethetechnicalrequirementofamarketingapplicationapprovalamongdifferentcountries/regions,bothindustryandregulatoryagencyallrealizedthatregulatorybodiesneedtoimprovethequality,efficiency,clarityandtransparencyofreviewtomarketingapplication.Recentyear,USFDAtriedtointroduceadocumentedbestpractice,GoodReviewPractice(GRP)tomeetthechallenges.Ithasbeenawardedbybothindustryandpublicuponimplementation.Inthispaper,authorstudiedUSFDA’sGoodReviewPractice(GRP)&itsderivativeregulationsaswellasitssignificanceafterimplementation,andtriedtodiagnoseandanalyzetheexistedreviewqualityandreviewmanagementrelatedissuesofChinaregulatorybody(CenterforDrugEvaluation,StateFoodDrugAdministration)throughcasesstudieswhichIencounteredpersonallyorcollectedfrompublicdomainofSFDAandCDE.WhilerefertoUSFDA’sGoodReviewPractice(GRP)andderivativeregulations,withtheconsiderationtocurrentsituationofbothChinagovernmentandindustry,itisclearlysuggestedthenecessityandimplicationtoestablish“GoodRegistration&ReviewPracticeinChina.ToprovidemyproposalforbuildingChinaregulatorybodywithstandardreviewprocedure,highquality,efficiency,clarityandtransparencyofreview,aconceptualChineseGoodRegistration&ReviewPractice(GRRP)isprovided.ItisalsoforeseenthatGRRPinChinawillalsogreatlybenefitChinapharmaceuticalindustryforguidingdrugdevelopment,andhelpusrealizethetruly-innovationthroughinternationalrecognizedprocedure.Keyword:review;regulatoryagency;practice;quality;1目录第1章序言1第2章发达国家"药品审评质量管理规范(GRP)"简介32.1美国FDAGRP的制订背景、内容、实施及其影响32.1.1.美国FDAGRP的制定背景32.1.2.美国FDAGRP的内容以及“药品审评质量管理原则和规范(GoodReviewManagementPrincipleandPractice,GRMP)”简介42.1.3美国FDAGRPs和GRMPs的实施及其影响142.2日本“药品审评质量管理规范”的制订背景、内容和实施简介15第3章我国药品审评和审批的现状173.1现有的“审评和审批”程序和法规简介173.1.1现行的药品审评和审批程序173.1.2现行药品审评和审批程序的配套法规和规章213.2存在的问题和挑战233.2.1关于公开233.2.2关于公平和公正283.2.3关于审评管理和审评质量313.2.4关于审评规范的实施33第4章在我国建立和实施“药品审评和审批质量管理规范(GRRP)”的必要性和建议354.1在我国建立和实施GRRP的必要性354.2对我国建立和实施GRRP的建议354.2.1.GRRP的框架354.2.2监督机制的建立和作用36第5章讨论385.1充足的人力资源是实现药品公开、公平和公正的必要条件385.2我国注册法规体系的机构改革势在必行405.3其他41第6章参考文献42第7章缩略语43致谢错误!未定义书签。1第1章序言在全球范围内,为了最大程度地保护和促进公众健康,制药行业都是一个受各国政府高度监管的行业。根据世界卫生组织(WHO)的定义,药品监管体制(DrugRegulatoryRegime)的主要目标在于政府部门根据具体的标准对药品生产、采购、进出口、流通、供应、售卖、广告以及临床试验等环节进行管理,以便确保药品的质量、安全及有效和产品信息的准确性[1]。从内容上看,一个国家的药品监管系统既包括药品上市之前的药品审评和审批(注册批准),也包括药品上市之后的风险再评估、广告监管和后继的研究,还涵盖了药品生产和流通环节中的各种质量认证与执法行为。而其中的重中之重,莫过于上市之前的药品审评和审批(注册)制度。无论从保障公共用药安全,还是从有效维护制药企业的合法商业利益出发,药品注册制度都发挥着至关重要的核心作用。世界卫生组织还在其报告中指出,药品注册制度(drugregistrationsystem)是一个国家药物政策和监管的最重要组成部分之一,而药品注册部门一般也是药品监管部门中的核心机构。药品注册从内容上看包括已有标准的化学药品审评、含有新化学物质的药品审评、传统药和草药的审评、临床试验的授权等;从程序上则涵盖了受理程序、基础审评和审批阶段、完全注册(包括药品上市后监测以及定期评估)以及药品再注册等环节[2]。包括我国在内的世界各国都对药品的上市准入采取许可制度。绝大部分国家需要对企业所提交的上市申请进行独立的技术审评和审批。在药品研发和上市批准的过程中,药品监管机构作为行政执法者,拥有最高的行政权力。工业界和研发机构作为行政相对人,一方面希望药品监管机构通过发布各类技术指导原则明确申请获批准所需的数据要求,另一方面也希望药品监管机构能够规范其审评或审批的行为。虽然国际人用药品注册技术要求国际协调会(ICH)的成立和推广极大程度上地统一了各国对药品在不同地区/国家申请批准时所需的数据要求,但在全球范围内,无论是工业界还是药品监管机构都意识到,监管机构在对上市申请的审评质量、审评效率、审评结果的明确性/可预见性和透明度方面都有待提高。近年来,美国食品药品监督管理局(FDA)率先试图通过建立“药品审评质量2管理规范(GRP)”及配套法规,以应对这样的挑战,并取得了良好的成效。“药品审评质量管理规范(GRP)”及配套法规是美国FDA下属“药品审评和研究中心”(CDER)内部讨论药品审评在程序、形式、内容和审评管理方面行为规范的一系列标准程序的集合,虽然它是FDA内部规章制度,但对社会公开。它的适用对象不仅是参与药品审评的所有审评及其审评管理人员,而且包括工业界和申请者。本论文通过对美国FDA已建立的GRP及配套法规,以及这些法规实施后产生的影响的介绍,结合本企业在我国药品申报和注册中观察到/遇到的一些现象/案例、国家药品监督管理局(SFDA)和药品审评中心(CDE)公开发布的一些数据,试图对我国药品监管机构(SFDA及其下属的CDE)在审评质量、审评管理规范性方面存在的问题和挑战进行诊断和分析。结合我国国情以及行业特点,参照国外药品监管机构已制订的“药品审评质量管理规范”以及细则,展望在我国建立“药品审评和审批质量管理规范(GRRP)”的必要性和内涵,并提出具体的框架。旨在为我国的药品审评/审批建设规范的审评程序、提高审评质量,效率,可预见性和透明度见言献策。同时,“药品审评和审批质量管理规范”在我国的建立也将为工业界的药品研发提供明确的指南,促使我国的新药研发尽早与发达国家接轨,实现真正地自主创新。基于此,本论文将包括以下主要内容:对发达国家“药品审评质量管理规范”制定背景、内容和实施情况进行介绍,主要反映了美国FDA当前的实践;对我国药品审评和审批程序和法规的介绍,以及对目前在审评审批过程中观察到的问题的分析和判断;探讨在我国制定“药品审评和审批质量管理规范”的必要性,以及提出初步的原则和设想最后,对我国药品注册体系存在的一些深层次的问题作一定的讨论。本文所持的观点和分析谨代表作者个人的观点,与本人所服务的公司无关。本人在论文中的第三
 三七文档所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
三七文档所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
 扫描二维码
扫描二维码








 alalalzzz
alalalzzz
本文标题:我国建立药品审评和审批质量管理规范的必要性和内涵
链接地址:https://www.777doc.com/doc-363145 .html