您好,欢迎访问三七文档
当前位置:首页 > 医学/心理学 > 药学 > 新药临床研究的例数估算问题
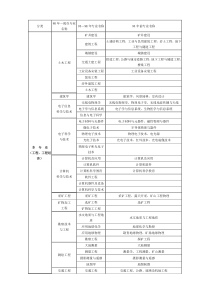
新药临床研究的例数估算问题medstar国家药品监督管理局最近对新药临床研究所需病例数做了规定。按统计学中样本例数的估算方法,并不能适应新药临床研究的需要,因为临床研究的目的不仅要分析新药疗效,而且要了解新药的不良反应,所以必须有一定的基本例数,才能满足科研中重复原则的要求。1新药临床研究的基本例数要求国家药品监督管理局规定临床研究的完成例数,见表1。表1新药临床研究所需的病例数临床研究分期化学药中药(试验组(例)盲法(对)开放(例)临床试验Ⅰ期/20~3020~30Ⅱ期≥100/≥100Ⅲ期≥300(试验组)≥300Ⅳ期/2000≥2000等效性试验临床试验≥60//生物利用度18~24//同时规定:①新药临床研究应完成符合统计学要求的临床病例数;②应在临床药理基地中选择负责和承担单位,并经核准,如需增加承担单位或在基地以外的医疗机构进行临床研究,须另行申请并经批准;③临床研究可进行多中心临床试验,每个中心的病例数不得少于20例;④第一类新药(化学药)中的避孕药,Ⅱ期临床试验应完成不少于100对6个月经周期的随机对照试验,Ⅲ期临床试验完成不少于1000对12个月经周期的开放试验,Ⅳ期临床试验应充分考虑该类药品的可变因素,完成足够样本量的研究工作。我国现将临床试验分为4期,即将原来的Ⅱ期分为Ⅱ、Ⅲ期。Ⅱ期采用随机盲法对照临床试验,着重于新药有效性的判断,并对安全性作初步评价,推荐临床给药剂量。Ⅲ期临床试验采用扩大的多中心临床试验,在进一步评价有效性的同时,着重于安全性的评价,因此对临床研究所需的病例数也做了相应的规定。首先,新药临床研究不得少于以上规定的最低例数。从有效性方面看,若新药的有效率远大于对照药,统计学上进行例数估算,不需按新药临床研究规定的最低例数即可得到P<0.05的结论,但进行例数估算时,代入公式计算的新药有效率是一估计值;另外,计算出的病例数只有理论意义,再加上病情、病种、病程、医院等诸多因素的影响,若按理论估算的病例数进行临床研究,未必得出预期的有效率和P<0.05的结论。从不良反应方面看,Ⅲ期临床试验中若新药的主要不良反应发生率为1%,则理论上100例患者中有1例发生,实际试验中有可能观察不到,如按规定的最低例数做300例,则临床试验中有80%的把握度能观察到此不良反应。同样,Ⅱ期临床做100例,则有80%的把握度能观察到发生率约为3%的常见的不良反应;Ⅳ期临床做2000例,则有80%的把握度能观察到发生率约为0.1%的少见的不良反应。其次,新药临床研究还应符合统计学要求。若试验误差较大或两组有效率很接近时,按表中的最低数进行试验,则可能达不到显著水平,所以临床医师应掌握病例数的估算方法,即根据统计学方法进行理论估算,再结合临床确定合适的研究例数。临床研究进行例数估算时,常取α=0.05,β=0.20。α是显著性水平,也是假阳性率,α=0.05表示将来自同一总体的两样本误认为来自不同总体的概率为5%。(1-β)称为检验效能把握度,β=0.20表示当两总体确有差别,按α水准有80%的把握能发现它们有差别。下文进行例数估算所使用之表格,均是按α=0.05,β=0.20所估算,新药临床研究所需最低例数的规定,也是以这些表格和计算为依据的。需要说明的是,表1盲法试验中病例数指的是“对”,如Ⅱ期临床试验,盲法“≥100”是指≥100对,即200例。此处指平行设计或是分层设计,其目的是加强可比性,更好地均衡。它与统计学上的配对设计(自身左右配对,自身前后配对或挛生配对)不同。后者一般在局部外用药、慢性疾病用药或抗癌药的研究及某些特殊情况如生物等效性检验中使用。由于试验性质不同,统计方法、例数估算方法也不同。2有效率显著性检验的例数估算这是临床研究中最常用的设计类型。对于平行设计或分层设计,在初步了解甲药有效率P1及乙药有效率P2后,按下式估算出每组应取多少例,即可有80%把握度,得到P<0.05的有显著性意义的统计结果。其基本公式是:式中P=(P1+P2)/2,Q=1-P,Uα=1.96,Uβ=0.842,故(Uα+Uβ)2=7.85,上式可简化为:统计书籍中用更精密的计算求出数据,列成表格,使用时应先算出有效率之差(D=P2-P1)及较小的发生率(设P2>P1,如P1<50%则L=P1,如P1>50%则L=100-P2),见表2。表2平行设计的例数估算表两率之差(%)D=P2-P15101520253035404550较小率(%)54201306944312420161412L=P1106801959659413023191613或1591025012071483426211714L=100-P220109029013580533828221815251250330150885740302319153013803601609360423123191535147038017096614231231814401530390175976142302217124515603901759660402821165015603901709357382619摘自郭祖超主编.医用数理统计方法[1]例1预期对照药有效率为80%,新药有效率为90%,以较小率为P1,故P1=0.8,P2=0.9,可算出:N1=N2=3.92×(1-(0.9+0.8-1)2)/(0.9-0.8)2=199.9(200例),应当各做200例,共需400例,才能以80%把握度得到P0.05的显著性水平;查表法D=90-80=10,P150,L=100-90=10,查出N=195,与公式所得基本相同。例2设P1=0.7,P2=0.9,则N1=N2=3.92×(1-(0.9+0.7-1)2)/(0.9-0.7)2=62.7≈63(例),100例,按规定仍应各做100例。查表法D=90-70=20,P150,L=100-90=10,查出N=59,与公式所得大致相近。从表中可以看出:当两率之差D在大于20%时,按新药要求做100对即可,不必进行估算。但在两率之差15%时,就一定要进行估算。国家药品监督管理局临床试验最低数规定的依据也在于此。当两组例数不等时,设新药与对照药比例取a∶b=3∶2,则按上表或公式算出N后,一组用Na=N×(a+b)/(2×b),另一组Nb=N×(a+b)/(2×a)进行计算。例1中,如按a∶b=3∶2,则Na=59×5/4=74例,Nb=59×5/6=49例。3配对设计中有效率显著性检验的例数估算配对设计主要指自身左右配对、自身前后配对,即同一患者用2种药物,或挛生配对,或严格的异体配对,后者应将多种因素考虑在内,配对的患者有严格的相同性,故试验实施较困难,现较少采用,本文仅做一简介。统计分析时应比较甲药优于乙药的百分率和乙药优于甲药的百分率,而不是比较甲药有效率和乙药有效率。所以,例数估算时不仅要知道P1及P2,还应知道两药均有效的百分率Pa,例数估算公式:式中B=(P1-Pa)/100,C=(P2-Pa)/100统计书籍中也有表格,方法同前,使用时先计算有效率之差(D=P2-P1)及较小率减两组均阳性的率(L=P1-Pa)查表,当P1-Pa>50时,改用L=100-(P2-Pa)进行查表。4生物等效性检验的例数估算我国将4类新药分为8亚类,其中有7类均可先做药品的生物利用度试验,若结果合格可免做临床验证试验,因此生物等效性检验的使用日益广泛。I期临床的生物等效性检验规定用交叉配对设计,即一组患者先用参比药,再用供试药(称为ROT组);另一组患者先用供试药,再用参比药(称为TOR组),两组患者人数相等,分配到何组也按随机原则进行。它属于狭义的配对设计,但并非差异性检验(配对差值统计分析),而是等效性检验。生物等效性检验是指新药的生物利用度与参比品相比,在低侧不低于参比品的80%,且应有单侧检验显著意义;在高侧不得高于参比品的125%,也应有单侧检验显著意义。因此,这种检验又称为“双向单侧T检验”。只有两个T值都是P0.05(有显著意义),才说明新药在生物利用度上,是与参比品等效的。生物利用度中吸收程度用“AUC(曲线下面积)的对数值之均数”进行对比,公式为:对R=1时:对R≠1时:其中σ取该药AUC的个体差异(CV%,即标准差均数之比值),δ是供试品与高限(或低限)之差,见表3。表3生物等效性检验的例数估算表比值R=MT/MR0.850.900.9511.051.101.151.20变异系数CV(%)5.012644468227.52286668124410.0361286810207612.5541610810143011815.07822121012204216817.510430161416265622620.013438201618327229422.516846242024409036825.0206562824284811045227.5248683428345813254430.02927040323868156642摘自DieterHauschke,etal.表中的R=MT/MR,即供试品与参比品均数之比值。由于用了对数值,故R=0.9的数据与R=1.1的数据不尽相同(ln(0.9)=-0.10536,ln(1.1)=0.09531,而ln(1/0.9)=ln(1.1111)=0.10535)。表中的变异系统(CV%),也即估计的“标准差/均数”,应根据既往数据或经验决定,一般制剂CV约为10%~20%,由表中可看出,当CV10%,R1±0.1或CV15%,R1±0.05时,取12例即可;超过该范围则需较多例数才能以80%把握度达到双向单侧T检验P0.05的要求。对于缓释制剂因CV较大,约为15%~25%,由表中可看出,当CV<15%,R<1±0.1或CV<20%,R<1±0.05时,约需18~24例。可见,国家药品监督管理局现规定不少于18~24例是合适的。
 三七文档所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
三七文档所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
 扫描二维码
扫描二维码









 魔剑旋
魔剑旋
本文标题:新药临床研究的例数估算问题
链接地址:https://www.777doc.com/doc-364517 .html