您好,欢迎访问三七文档
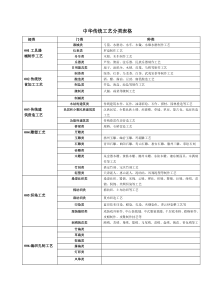
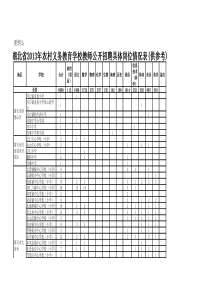
名词解释2.被动转运:是指存在于膜两侧的药物服从浓度梯度扩散的过程,分为单纯扩散和膜孔转运两种形式。3.溶出速率:是指在一定溶出条件下,单位时间药物溶解的量。4.载体媒介转运:借助生物膜上的载体蛋白作用,使药物透过生物膜而被吸收的过程,可分为促进扩散和主动转运两种形式。6.ATP驱动泵:以ATP水解释放的能量为能源进行主动转运的载体蛋白家族。7.生物药剂学:是研究药物及其剂型在体内的吸收、分布、代谢与排泄过程,阐明药物的剂型因素、机体的生物因素与药理效应三者之间相互关系的科学。8.吸收(Absorption):药物从用药部位进入体循环的过程。分布(Distribution):药物进入体循环后向各组织、器官或者体液转运的过程。代谢(Motabolism):药物在吸收过程或进入体循环后,受肠道菌丛或体内酶系统的作用,结构发生转变的过程。排泄(Excretion):药物或其代谢产物排出体外的过程。转运(transport):药物的吸收、分布和排泄过程统称为转运。处置(disposition):分布、代谢和排泄过程称为处置。消除(elimination):代谢与排泄过程药物被清除,合称为消除。9.细胞通道转运:药物借助其脂溶性或膜内蛋白的载体作用,穿过细胞而被吸收的过程。10.细胞旁路通道转运:是指一些小分子物质通过细胞间连接处的微孔进入体循环的过程11.载体媒介转运(carrier-mediatedtransport)借助生物膜上的载体蛋白作用,使药物透过生物膜而被吸收的过程12.促进扩散又称易化扩散,是指某些非脂溶性药物也可以从高浓度处向低浓度处扩散,且不消耗能量。13.pH-分配假说:药物的吸收取决于药物在胃肠道中的解离状态和油/水分配系数。14.蓄积:某些药物接续应用时,在机体的某些组织中的药物有浓度逐渐上升的趋势。15.首过效应:药物在消化道和肝脏中发生的生物转化作用,使部分药物被代谢,最终进入体循环的原型药物量减少的现象。16.肾清除率:肾清除率代表在一定时间内(通常以每分钟为单位)肾能使多少积(通常以毫升为单位)的血浆中该药物清除的能力,如肾对某药物清除能力强时表示有较交多血浆中的该药物被清除掉。17.肾小球滤过率:单位时间内(每分钟)两肾生成的超滤液量称为肾小球滤过率。18.双峰现象:某些药物因肠肝循环可出现第二个血药浓度高峰,该现象称为双峰观象。19.肠肝循环:在胆汁中排泄的药物或其代谢物在小肠中移动期间重新被吸收返肝门静脉血的现象。20.药物动力学,是应用动力学原理与数学处理方法,研究药物通过各种途径(如静脉注射、静脉滴注、口服等)给药后在体内的吸收、分布、代谢、排泄过程(即ADME过程)量变规律的学科,致力于用数学表达式阐明不同部位药物浓度(数量)与时间之间的关系。答题:1.简述促进扩散的特点,并与被动转运比较两者的异同。促进扩散是指某些物质在细胞膜载体酌帮助下,由膜高浓度侧向低浓度侧扩散的过程。其特点有:①药物从高浓度侧向低浓度侧的顺浓度梯度转运;②需要载体参与,载体物质通常与药物有高度的选择性;③不消耗能量,扩散过程与细胞代谢无关,不受细胞代谢抑制剂的影响;④转运有饱和现象;⑤结构类似物能产生竞争性抑制作用;⑥有结构特异性和部位特异性。与被动转运相同的是:促进扩散服从顺浓度梯度扩散原则,不消耗能量。与被动转运不同的是:由于载体参与,促进扩散的速度要比单纯扩散的速度快得多。2.简述主动转运的分类及特点。借助载体或酶促系统的作用,药物从膜的低浓度侧向高浓度侧的转运称为主动转运。主动转运可分为ATP驱动泵和协同转运两种。ATP驱动泵是以ATP水解释放的能量为能源进行主动转运的载体蛋白家族。协同转运是依赖另一种物质的电化学梯度所贮存的能量对物质进行主动转运,而维持这种电化学势是钠钾泵或质子泵。主动转运的特点有:①逆浓度梯度转运;②需要消耗机体能量;③需要载体参与,载体物质通常与药物有高度的选择性;④主动转运的速率及转运量与载体的量及其活性有关,具有饱和作用;⑤结构类似物能产生竞争性抑制作用,相似物竞争载体结合位点,影响药物的转运;⑥受代谢抑制剂的影晌;⑦有结构特异性和部位特异性。3.简述生物药剂学中讨论的生理因素对药物吸收的影响。(1)消化系统因素:①胃肠液的成分与性质:胃液的pH呈酸性,有利于弱酸性药物的吸收;小肠较高的pH环境是弱碱性药物最佳的吸收部位。②胃排空和胃空速率:胃排空速率慢,药物在胃中停留时间延长,弱酸性药物吸收会增加,但是胃排空加快,到达小肠部位所需的时间缩短,有利于药物在小肠部位吸收。③小肠内运行:可促进固体制剂进一步崩解、分散,使之与肠分泌液充分混合,增加了药物与肠表面上皮的面积,有利于难溶性药物的吸收。④食物的影响:食物不仅能改变胃空速率而影响吸收,也可能促进药物的吸收或不影响吸收。⑤胃肠道代谢作用的影响:药物的胃肠道代谢是一种首过效应,对药物疗效有一定的甚至很大的影响。(2)循环系统因素:①胃肠血流速度:血流量可明显影响胃的吸收速度,但这种现象在小肠吸收中不显著;②肝首过作用:肝首过效应愈大,药物被代谢越多,药效会受到明显的影响;③肠肝循环:对经胆汁排泄的药物有影响,可使药物的作用明显延长;④淋巴循环:对大分子药物的吸收起着重要作用。(3)疾病因素:胃酸缺乏、腹泻、甲状腺功能不足、部分或全部胃切除、肝脏疾病等均可影响药物胃肠道吸收。5.药物的溶出速率对吸收有何意义?影响其溶出速率的因素有哪些?药物的浴出是指药物从制剂中溶解到溶出介质中的过程。对难溶性药物而言,溶出是其吸收的限速过程,药物在胃肠道内的溶出速率直接影响药物的起效时间、药效强度和作用持续时间。影响药物溶出速率的因素主要有:①药物的溶解度,药物的溶解度与溶出速度直接相关,当药物在扩散层中的溶解度增大,则可加快药物的溶出速率。②粒子大小,药物粒子越小,则与体液的接触面积越大,药物的溶出速度增大,吸收也加快。③多晶型,不同的晶型溶解度不同,其溶出速度也不同,直接影响药物的吸收速度,并会影响药物的药理作用。④溶剂化物,将药物制成无水物或有机溶剂化物,有利于溶出和吸收。6.影响Ⅱ型药物口服吸收的理化因素有哪些?如何改善该类药物的口服生物利用度?Ⅱ类药物是低溶解性/高渗透性的一类药物,因药物在胃肠道溶出缓慢而限制了药物的吸收。影响Ⅱ类药物吸收的理化因素有药物的溶解度、晶型、溶媒化物、粒子大小等。增加药物的溶解度和(或)加快药物的溶出速率均可有效地提高该类药物的口服吸收,主要方法有:①制成可溶性盐类;②选择合适的晶型和溶媒化物;③加入适量表面活性剂;④用亲水性包合材料制成包合物;⑤增加药物的表面积;⑥增加药物在胃肠道内的滞目时间;⑦抑制外排转运及药物肠壁代谢。7.采用什么给药途径可避免肝首过效应?试结合各给药途径的生理学特点说明其避免首过效应的原理。可以通过改变给药途径尽量避免首过效应,尤其是肝首过效应。主要途径有:(1)薛脉、肌内注射:静脉注射直接进入体循环,因此不存在首过效应;肌内注射经毛细血管吸收进入体循环,不经门肝系统,因此不存在首过效应。(2)口腔黏膜吸收:口腔黏膜下有大量毛细血管汇总至颈内静脉,不经肝脏而直接进入心脏,可绕过肝脏的首过作用。一般可制成口腔贴片给药。(3)经皮吸收:药物应用到皮肤后,首先从制剂中释放到皮肤表面,溶解的药物分配进入角质层,扩散通过角质层到达活性表皮的界面,再分配进入水性的活性表皮,继续扩散到达真皮,被毛细血管吸收进入血液循环,可避开门肝系统。(4)经鼻给药:鼻黏膜血管丰富,鼻黏膜渗透性高,有利于全身吸收。药物吸收后直接进入体循环,无首过效应。(5)经肺吸收:肺泡表面积大,具有丰富的毛细血管和极小的转运距离,因此肺部给药吸收迅速,而且吸收后药物直接进入血液循环,不受肝首过效应的影响。(6)直肠给药:栓剂距肛门2cm处,可使大部分药物避开肝首过效应,给药生物利用度远高于距肛门4cm处给药。当栓剂距肛门6cm处给药时,大部分药物经直肠上静脉进入门静脉--肝脏系统。淋巴循环也有助于直肠药物吸收,经淋巴吸收的药物可避开肝脏代谢作用。8、决定药物被组织摄取和积蓄的主要因素是什么?答:影响药物被组织摄取和积蓄的主要因素是组织器官的血液灌流速度和药物与组织器官的亲和力。而药物与组织器官的亲和力主要和药物的结构、解离度、脂溶性以及蛋白质结合率有关。通常血流丰富的组织蛇舞药物的速度快。2、表观分布容积的意义答:①Vd值它代表药物透膜转运和分布到体内各部位的特性。是由药物的理化性质决定的常数。②Vd=D/C反映药物剂量与血药浓度的关系,利用此公式,若测得血药浓度,乘以其表观分布容积,即可求得药物在体内的总量。对指导临床用药具有重要意义。4.药物血浆蛋白结合和组织蛋白结合对表观分布容积和药物消除有何影响?当药物主要与血浆蛋白结合时,其表观分布容积小于它们的真实分布容积;而当药物主要与血管外的组织结合时,其表观分布容积大于它们的真实分布容积。蛋白结合率高的药物,通常体内消除较慢。5.讨论药物蛋白结合率的临床意义,药物蛋白结合率的轻微改变(如变化率为1%),是否会显著影响作用强度?药物与血浆蛋白结合,能降低药物的分布与消除速度,延长作用时间,并有减毒和保护机体的作用。若药物与血浆蛋白结合率很高,药理作用将受到显著影响。由于药理作用主要与血中游离药物浓度有关,因此血中游离药物浓度的变化是影响药效的重要因素。对于高蛋白结合率的药物,药物蛋白结合率的轻微变化,可能导致血中游离药物浓度明显变化,因而会显著影响药物作用强度。如结合率高达98%的药物,若结合率降低1%,则可以使游离药物浓度上升1.5倍,可显著影响作用强度。对于低蛋白结合率的药物,结合率的轻微改变,对血中游离药物浓度影响不明显因此不会显著影响药物作用强度。6、提高药物脑内分布的方法答:①、颈动脉灌注高渗甘露醇溶液,使血脑屏障暂时打开,增加药物入脑②、对药物结构进行改造,引入亲脂性基团,制成前药,增加化合物脂溶性③、使用聚氰基丙烯酸酯、聚乳酸、乳酸—羟基乙酸共聚物等高分子材料,将药物装载制成纳米粒,可提高药物的脑内分布④、利用脑毛细血管内皮细胞上存在的特异性载体⑤、通过鼻腔途径给药,可以使药物绕过血脑屏障,直接进入脑组织9.为什么微粒在体内的半衰期很短,如何延长微粒在血液中循环时间?常规设计的微粒给药系统在体内会被网状内皮系统的单核巨噬细胞吞噬,因此半衰期很短。通过改善微粒的亲水性,增加微粒的柔韧性及其空间位阻,干扰吞噬细胞对微粒的识别过程,则可明显延长微粒在血液中的循环时间。目前最常用的方法就是采用表面修饰技术,该技术系通过一定的化学反应,将非离子型聚合物如聚乙二醇(PEG)以共价结合的方式引入到微粒的表面,既提高了微粒的亲水性和柔韧性,又明显增加微粒的空间位阻,使微粒具有隐蔽性,不易被单核巨噬细胞识别和吞噬,从而达到长循环的目的。10.影响微粒给药系统体内分布的因素有哪些?(1)细胞和微粒之间的相互作用,包括内吞作用、吸附作用、融合作用、膜间作用等。(2)微粒本身的理化性质,包括粒径、电荷、表面性质的影响。(3)微粒的生物降解。(4)机体的病理生理状况。1.药物代谢对药理作用可能产生哪些影响?①使药物失去活性,代谢可以使药物作用钝化,即由活性药物变化为无活性的代谢物,使药物失去治疗活性;②使药物活性降低,药物经代谢后,其代谢物活性明显下降,但仍具有一定的药理作用;③使药物活性增强,即药物经代谢后,表现出药理效应增强,有些药物的代谢产物比其原药的药理作用更强;④使药理作用激活,有一些药物本身没有药理活性,在体内经代谢后产生有活性的代谢产物,通常的前体药物就是根据此作用设计的,即将活性药物衍生化成药理惰性物质,但该惰性物质能够在体内经化学反应或酶反应,使母体药物再生而发挥治疗作用;⑤代谢产生毒性代谢物,有些药物经代谢后可产生毒性物质。2.简述药物代谢反应的类型。通常可以分为I相反应和Ⅱ相反应两大类。I相反应包括氧化反应、还原反应和水解反应,参与各种不同反应的药物代谢酶依次为氧化酶、还原酶和水解酶,通常是脂溶性较强的药物通过反应生成极性基团。Ⅱ相反应即结合反应,参与
 三七文档所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
三七文档所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
 扫描二维码
扫描二维码








 dwyct
dwyct
本文标题:生物药剂学背诵
链接地址:https://www.777doc.com/doc-2201356 .html