您好,欢迎访问三七文档
当前位置:首页 > 商业/管理/HR > 广告经营 > crispr-cas9进行全基因组的筛选
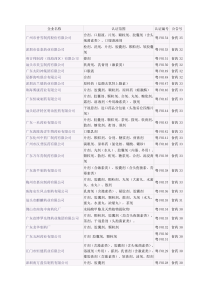
HumanCRISPRKnockoutPooledLibrary(Brunello)一、lentiCas9-Blast质粒的基本信息:由于cas9自待的抗性基因是我们不需要,在抗性基因两边突变出酶切位点,酶切掉原本的抗性基因。换成另一种。1.lentiCas9-Blast质粒购买来时是以细菌的形式存在的,先需要扩大培养。2.试剂盒提取质粒,并测定浓度。二、慢病毒包装质粒1转染前24h,用胰蛋白酶消化对数生长期的HEK293T细胞,调整细胞为7*105在5ml的DMEM培养基中。37℃、5%CO2培养箱内培养。待细胞密度达70%~80%时即可用于转染。2转染前2h将细胞培养基更换为无血清培养基。3向离心管中加入质粒DNA和等体积的Opti-MEM,混匀,调整总体积为2.5ml,在室温下温育5分钟。4将Lipofectamine2000试剂轻柔摇匀,取100μlLipofectamine2000试剂在另一管中与2.4mlOpti-MEM混合,在室温下温育5分钟。5把稀释后的DNA与稀释后的Lipofectamine2000进行混合,轻轻地颠倒混匀,不要振荡。必须在5分钟之内混合。6混合后,在室温下温育20分钟,以便形成DNA与Lipofectamine2000稀释液的转染复合物。7DNA与Lipofectamine2000混合液转移至293T细胞的培养液中,混匀,于37℃,5%CO2细胞培养箱中培养。8培养8h后倒去含有转染混和物的培养基,每瓶细胞加入20ml的PBS液,轻轻左右晃动一下培养瓶以洗涤残余的转染混和物,然后倒去。9每瓶细胞中加入含10%血清的细胞培养基25ml,于37℃、5%CO2培养箱内继续培养48小时。10收集转染后48小时(转染即可为0小时计起)的293T细胞上清液。11于4℃,4000g离心10min,除去细胞碎片。12以0.45μm滤器过滤上清液于15ml离心管中。三、慢病毒转染细胞参考网址:取六孔板铺50000个细胞,37°C,5%CO2培养,待细胞长到70%,换含有8ug/mlpolybrene的培养基培养。2.慢病毒悬液用含有10ug/ml的DMEM培养基稀释。3.六孔板中每个空加一种浓度的病毒稀释液,每空0.5ml。4.孵育48~72小时。5.换成含有适宜浓度的抗生素的培养基培养1.5ml,筛选出转染成功的细胞。抗生素浓度的摸索:用培养基稀释抗生素为1ug/ml、2ug/ml、3ug/ml、4ug/ml、5ug/ml、6ug/ml、7ug/ml、8ug/ml、9ug/ml、10ug/ml,并用稀释后的浓度培养细胞,3~5天内使所有细胞死亡的最低浓度可用于筛选细胞。6.持续加药筛选直到细胞不再大量,并能稳定表达Cas9。一般筛选需要1~2个星期。7.在转导sgRNA文库前2~7天改用正常培养基培养。(或者先测定MOI值后,根据MOI加病毒的量,操作可参照sgRNA的转导)四、检测病毒转染的结果1.试剂盒提取质粒2.PCR扩增()。引物:ForwardPrimer:dCas9-F2CCAAAGAGGTGCTGGACGReversePrimer:Blast-RGCTCTTTCAATGAGGGTGGA3.2%琼脂糖凝胶电泳检测,并用试剂盒纯化PCR产物4.通过免疫印迹扩增,收集,裂解并测试Cas9表达的细胞群。五、lentiGuide-Puro骨架基本信息六、SgRNAlibrary的设计:在基因芯片上合成核苷酸,分离出来后,进行PCR扩增。七、把设计好的sgRNA整合到质粒上:)SgRNA文库的构建1,lentiGuide-Puro载体可以使用BsmBI和一对退火的寡核苷酸可以把sgRNA克隆到骨架中。首先把5ug的lentiviralCRISPR质粒用BsmBI混合37度,酶切30min。2,试剂盒(QIAquickGelExtractionKit)纯化酶切的质粒,并用EB或ddH2O溶解。3,sgRNA1和sgRNA2用T4链接酶连接。4,反应程序:5,把第三步的产物用无菌水或EB以1:200的比例稀释。6,质粒与sgRNA的连接整合,温室放置10min。7.异丙醇沉淀核酸,75%乙醇洗涤后,晾干,用ddH2O溶解。8.测质粒DNA的浓度。八、sgRNA文库的扩增质粒转化电感细胞试剂:•100µLelectrocompetentcells(STBL4TM,ThermoFisherScientific,11635-018)•400nglibraryplasmidDNA•4electroporationcuvettes(0.1cmgap,Bio-Rad,165-2089)•10mLSOC(1XSOC,NewEnglandBioLabs,B9020S)•4bioassayplates(500cm2,LBagar+antibiotic)•2Maxi-preps(QiagenHiSpeedMaxi,12663)•Biospreader(BactiCellSpreader,VWRInternational,60828-684)•Electroporator(MicroPulserTM,Bio-Rad,16521001.37度预热SOC培养基15min和含有100ug/mlAMP的LB培养基。2.取电转化感受态细胞于冰上解冻,,标准质粒也置于冰上,同时准备干净的电击杯于冰上预冷。3.取400ng的质粒加入100ul的电感细胞中,充分混匀。4.取25ul混合液把电击杯放在电击仪上进行电击(1.8kv)。5.迅速在电击杯中加入1mlSOC培养液(无抗生素),然后充分混匀,移到15ml的离心管中.6.重复3次,增加SOC培养基到10ml。7.取两空离心管,分别加入5ml上述混合液。8.37度放置1h。9.取10ul的混合物做4个浓度梯度,每个稀释10倍。10.每个浓度(稀释103、104、105、106)取10ul涂布于含有100ug/mlAmp的LB培养基。11.倒置培养皿,37℃培养16-18小时。12.计算电转的效率,菌落数*稀释倍数*电感细胞数。13.第二天刮取菌落接种含有50mlLB和100ug/mlAmp培养基的锥形瓶培养,摇床37度培养过夜。九、检测扩增的质粒1.用试剂盒提取质粒,按说明书操作。并测浓度。2.双酶切质粒3.用1%琼脂糖凝胶电泳检测,分析电泳后的条带的大小。十、慢病毒包装质粒1转染前24h,用胰蛋白酶消化对数生长期的293T细胞,以含10%血清的培养基调整细胞密度为1.2x107细胞/20ml,重新接种于15cm细胞培养皿,37℃、5%CO2培养箱内培养。24h待细胞密度达70%~80%时即可用于转染2转染前2h将细胞培养基更换为无血清培养基。3向离心管中加入质粒DNA和等体积的Opti-MEM,混匀,调整总体积为2.5ml,在室温下温育5分钟。4将Lipofectamine2000试剂轻柔摇匀,取100μlLipofectamine2000试剂在另一管中与2.4mlOpti-MEM混合,在室温下温育5分钟。5把稀释后的DNA与稀释后的Lipofectamine2000进行混合,轻轻地颠倒混匀,不要振荡。必须在5分钟之内混合。6混合后,在室温下温育20分钟,以便形成DNA与Lipofectamine2000稀释液的转染复合物。7DNA与Lipofectamine2000混合液转移至293T细胞的培养液中,混匀,于37℃,5%CO2细胞培养箱中培养。8培养8h后倒去含有转染混和物的培养基,每瓶细胞加入20ml的PBS液,轻轻左右晃动一下培养瓶以洗涤残余的转染混和物,然后倒去。9每瓶细胞中加入含10%血清的细胞培养基25ml,于37℃、5%CO2培养箱内继续培养48小时。10收集转染后48小时(转染即可为0小时计起)的293T细胞上清液。11于4℃,4000g离心10min,除去细胞碎片。12以0.45μm滤器过滤上清液于离心管中。或者:1用ddH2O溶解50mg的PEI,调PH为7.1,定容到50ml。2转染前24h,用胰蛋白酶消化对数生长期的HEK293T细胞,调整细胞为1.8*107在45ml的DMEM培养基中。37℃、5%CO2培养箱内培养。3取一个50ml离心管混合以下物质:45加入195ul的PEI试剂,常温放置10min。6再加入25mlDMEM。7跟换新的培养基848h后收集上清,0.45μm滤器过滤上清液于离心管中。十一、慢病毒文库转染模式细胞并进行基因筛选1.测定最适的MOI(1).测定前一天,取6孔板,每个孔加4×106个细胞,和2ml含有8ug/mldpolybrene的培养基。(2).测定病毒的浓度,准备7~10个无菌的Ep管。在每个管中加入90μl的无血清培养基。(3).取待测定的病毒原液10μl加入到第一个管中,混匀后,取10μl加入到第二个管中。继续相同的操作直到最后一管。(4).六孔板的每孔吸取90μl培养基丢弃。加入90μl稀释好的病毒溶液。放入培养箱培养。(5).24小时后,换成完全培养基2ml。小心操作,不要吹起细胞。(6).观察荧光感染后72小时后,于荧光显微镜下观察细胞状态和荧光情况,确定细胞状态较好且荧光数较多的孔,对应为较适宜的MOI值。MOI=病毒量/细胞量。(确定MOI范围a.带荧光标签的慢病毒:观察荧光感染后72小时后,于荧光显微镜下观察细胞状态和荧光情况,确定细胞状态较好且荧光数较多的孔,对应为较适宜的MOI值。b.不带荧光标签的慢病毒:通过抗性筛确定最适MOI值。慢病毒感染后72小时加入100ug/ml的AMP,培养3-5天,镜下观察,选取细胞存活率较高且细胞状态较好的孔,对应为较适宜的MOI值。)2.取病毒(最适的MOI值)感染已经稳定表达cas9的细胞系,于37℃、5%CO2培养箱内继续培养48小时,并同时转导未导入Cas9的细胞做对照。3.48h以后向细胞添加合适的选择剂量的嘌呤霉素。嘌呤霉素浓度的摸索:用培养基稀释嘌呤毒素为1ug/ml、2ug/ml、3ug/ml、4ug/ml、5ug/ml、6ug/ml、7ug/ml、8ug/ml、9ug/ml、10ug/ml,并用稀释后的浓度培养细胞,3~5天内使所有细胞死亡的最低浓度可用于筛选细胞。4.选择成功转导的细胞后,改成正常的完全培养基培养2~7天。(如果是crispr和cas9分开额双质粒系统,前面导入的就是携带cas9的质粒,带cas9稳定表达后,在这里后就能把构建好的sgRNA文库转染细胞,重复以后的步骤,MOI与加cas9时的相似,然后用相应的抗生素在筛选细胞。)5.提取筛选出来的细胞的基因组DNA和未转导sgRNA的细胞基因组DNA。7.基因组高通量测序和进行RIGER分析确定出几个高丰度的sgRNA,确定出目的基因。
 三七文档所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
三七文档所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
 扫描二维码
扫描二维码








 shaoguan001
shaoguan001
本文标题:crispr-cas9进行全基因组的筛选
链接地址:https://www.777doc.com/doc-4419887 .html