您好,欢迎访问三七文档
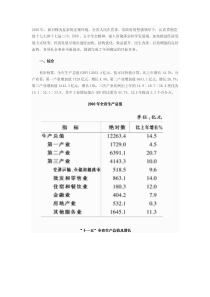
新药审批一新药的定义和注册分类(一)新药的定义原有定义:1999年《新药审批办法》“我国未生产的药品。已生产的药品①改变剂型、②改变给药途径的,增加新的适应症或制成新的复方制剂,亦按新药管理现行定义:2002年《药品管理法实施条例》“新药系指未曾在中国境内上市销售的药品。已生产的药品①改变剂型、②改变给药途径的,亦按新药管理(二)药品注册分类(附件)1、中药、天然药物(1)未在国内上市销售的从中药、天然药物中提取的有效成份及其制剂。(2)未在国内上市销售的来源于植物、动物、矿物等药用物质制成的制剂。(3)中药材的代用品。(4)未在国内上市销售的中药材新的药用部位制成的制剂。(5)未在国内上市销售的从中药、天然药物中提取的有效部位制成的制剂。(6)未在国内上市销售的由中药、天然药物制成的复方制剂。(7)未在国内上市销售的由中药、天然药物制成的注射剂。(8)改变国内已上市销售药品给药途径的制剂。(9)改变国内已上市销售药品剂型的制剂。(10)改变国内已上市销售药品工艺的制剂。2、化学药品1)未在国内外上市销售的药品:(1)通过合成或者半合成的方法制得的原料药及其制剂;(2)天然物质中提取或者通过发酵提取的新的有效单体及其制剂;(3)用拆分或者合成等方法制得的已知药物中的光学异构体及其制剂;(4)由已上市销售的多组份药物制备为较少组份的药物;(5)新的复方制剂。2)改变给药途径且尚未在国内外上市销售的制剂。3)已在国外上市销售但尚未在国内上市销售的药品:(1)已在国外上市销售的原料药及其制剂;(2)已在国外上市销售的复方制剂;(3)改变给药途径并已在国外上市销售的制剂。4)改变已上市销售盐类药物的酸根、碱基(或者金属元素),但不改变其药理作用的原料药及其制剂。5)改变国内已上市销售药品的剂型,但不改变给药途径的制剂。3、生物制品(1)治疗用生物制品(14类)(2)预防用生物制品(14类)有关术语定义1、药品注册:指依照法定程序,对拟上市销售的药品的安全性、有效性、质量可控性等进行系统评价,并作出是否同意进行药物临床研究、生产药品或者进口药品决定的审批过程,包括对申请变更药品批准证明文件及其附件中载明内容的审批2、药品注册申请人:指提出药品注册申请,承担相应法律责任,并在该申请获得批准后持有药品批准证明文件的机构。3、新药申请:未曾在中国境内上市销售药品的注册申请。已上市药品改变剂型、改变给药途径的,按照新药管理。4、药品注册申请:新药申请、已有国家标准药品申请、进口药品申请、补充申请统称药品注册申请(三)药品注册管理机构国家药品监督管理局(SFDA)主管全国药品注册管理工作,负责对药物临床研究、药品生产和进口的审批。省、自治区、直辖市药品监督管理局受国家药品监督管理局的委托,对药品注册申报资料的完整性、规范性和真实性进行审核。二、药品注册管理法制化的发展(一)20世纪上半叶,基本无药品注册管理制度近六十年发生的药物毒性的严重事件年代药品中毒事件1935~1937用二硝基酚减肥,引发白内障、骨髓抑制,死亡177人(美)1937磺胺醑剂(二甘醇)口服死170人(美)1937~1939用孕激素保胎,治先兆流产,女婴600多例生殖器官男性化(美)1954~1956有机锡(Stalinon)制剂治疗疮,死亡100人(法)1959~1961反应停(Thalidomide)灾难,畸胎出生2000~10000人,(德、西欧)“反应停”灾难(20世纪50年代和60年代)20世纪50年代,科学家推出一种新药,据说它能在妊娠期控制精神紧张,防止孕妇恶心,并且有安眠作用。这药名叫“反应停”(即酞胺哌啶酮——译注)。它由美国开发,1957年首次被用处方,后来在欧洲也可以买到。反应停的两种手性结构分子被反应停夺去胳膊的孩子们“反应停”灾难到了1960年,医生们对很多新生儿四肢缩短和其他畸形的善开始产生警觉。究其原因是孕妇服用了“反应停”。该药在1961年被禁用,但当时全世界约有8000名婴儿已经受害。经过很长一段时间法律的上的交锋,开发“反应停”的医药公司同意赔偿受害者的损失。“反应停”在出售之前,并未仔细检验其可能产生的副作用。“反应停儿童”的事件是一次惨痛的教训。它提醒人们,任何新药在用于临床之前必须经过彻底检验,尤其是用于孕妇的药物。并非每一位服用过“反应停”的母亲生育的都是残疾婴儿。但由于出现的概率是如此之高,其后果如此之惨痛,以至这种危险一旦为人们所知,该药便立即被停止出售。现在,医药公司对试验新药已倍加小心。(二)20世纪60年代开始将药品注册纳入法制化管理1、定义新药,明确药品注册范围;2、明确新药注册集中于中央政府卫生行政部门(或有关部门)专门机构负责审批注册;3、规定申请和审批程序;4、规定申请者必须提交的研究资料;5、制定各项试验研究指南;6、实行GLP(药物非临床实验质量管理规范)和GCP(药物临床实验质量管理规范)7、规定已在国外上市而未曾在本国上市进口药品,按新药对待(三)20世纪90年代药品注册管理的进展新药审评工作标准化、规范化发展建立了ICH(InternationalConferenceonHarmonizationofTechnicalforRegistrationofPharmaceuticalsforHumanUse)将新药的经济学研究列入注册规定范围《药品注册管理办法》2002年10月我国药品监督管理部门,国务院食品药品监督管理部门(以下简称SFDA)发布《药品注册管理办法》及其附件,于2002年12月1日实施。中国现行《药品注册管理》总则适应范围:在中国境内从事药物研究和临床研究,申请药物临床研究,药物生产或者进口,以及进行相关的药品注册检验监督管理。《药品注册管理办法》共18章208条新药注册管理《药品注册管理办法》中我们主要介绍药物的临床前研究,药物的临床研究,新药的申报与审批,新药的技术转让等。药物的临床前研究临床前研究内容:包括药物合成工艺,提取方法,理化性质及纯度,剂型选择,处方筛选,制备工艺,检验方法,质量指标,稳定性,药理毒理,动物药代动力学等。中药制剂还包括原材料的来源,加工及炮制等。生物制品还包括菌毒种,细胞株,生物组织等起始材料的质量标准,保存条件,遗传稳定性及免疫学的研究。临床前研究可概括为三方面1、综述研究2、药学研究:1制备工艺(合成、提取)、2理化性质、3纯度、4检验方法、5处方筛选、6剂型、7稳定性、8质量标准3、药理毒理研究1)主要药效学研究:2)一般药理研究:观察主要药效以外的其它作用3)毒理学研究(1)全身性用药的毒性试验(急性、长期)(2)局部用药的毒性试验(3)特殊毒理研究(致突变、生殖毒性、致癌)(4)药物依赖性试验4)药代动力学研究要求:安全性评价遵循《药物非临床研究质量管理规范》(GLP)新药研究开发的竞争与风险高投入一个有价值的新药,往往需要花费10-12年的时间,平均耗资大约在3.5亿美元左右高风险成功率低,即使已经进入临床试验阶段的试验性新药(IND)也有1/3—1/2要被淘汰出局新药(化合物)的开发过程产品监督临床试验(人体)临床前试验(动物)合成检验与筛选研究阶段产品介绍注册年12-55-1010-20化合物10,000-30,000化合物11211109876543210Source:PMAⅡ期Ⅲ期Ⅳ期Ⅰ期药物的临床研究药物临床研究必须经SFDA批准后实施。药物临床研究包括临床试验和生物等效性实验。临床研究必须执行《药物临床试验质量管理规范》。临床试验分为Ⅰ.Ⅱ.Ⅲ.Ⅳ期。Ⅰ期试验组人数为20-30例,Ⅱ期病例数为100例,Ⅲ期病例数为300例,Ⅳ期病例数为2000例。我国的新药临床试验分四期Ⅰ期临床试验是在人体进行新药研究的起始期,主要目的是研究人对新药的耐受程度,了解新药在人体内的药代动力学过程,提出新药安全有效的给药方案。Ⅱ期临床试验为随机盲法对照临床试验,由药物临床基地组织有条件的医院进行临床试验。其目的是确定药物的疗效适应证,了解药物的毒副反应,对该药的有效性安全性作出初步评价。Ⅲ期临床试验是Ⅱ期临床试验的延续,目的是在较大范围内进行新药疗效和安全性评价。要求在Ⅱ期临床试验的基础上除增加临床试验的病例数之外,还应扩大临床试验单位。多中心临床试验单位应在临床药理基地中选择,一般不少于3个。每个中心的病例数据不得少于20例。对此阶段的各项要求与Ⅱ期基本相似,但一般不要求双盲法。Ⅳ期临床试验也称上市后监察(postmarketingsuneillance)其目的在于进一步考查新药的安全有效性,即在新药上市后,临床广泛使用的最初阶段,对新药的疗效、适应证、不良反应、治疗方案可进一步扩大临床试验,以期对新药的临床应用价值做出进一步评价,进一步了解的疗效、适应证与不良反应情况,指导临床合理用药。Ⅳ期临床试验的内容应包括:①扩大临床试验②特殊对象临床试验:针对特殊人群(小儿、孕妇、哺乳期妇女、老人及肝肾功能不全的患者)的不同情况,设计临床试验方案③补充临床试验:重点是适应证的有效性观察或不良反应考察药物临床试验场所申请人应从具有药物临床试验资格的机构中选择进行临床试验临床研究用药制备和使用管理临床试验药物应当在符合GMP(药品生产质量管理规范)条件的车间,严格按照GMP要求制备、提供。申请人对临床研究用药的质量负有全部责任。临床研究药物使用由临床研究者负责,必须保证按研究方案使用于受试者;不得把药物交给任何非临床研究者。临床研究用药物不得销售。按时向药品监督管理部门报送资料临床研究方案及相关资料,应按规定在临床研究实施前报送SFDA和省级药品监督管理局。申请人完成每期临床试验后,应提交临床研究和统计分析报告。临床研究时间超过1年的,申请人每年都应提交研究进展报告。临床研究批文的有效时限药物临床研究被批准后应当在2年内实施,逾期作废,应当重新申请。保障受试者安全临床研究机构和临床研究者有义务采取必要措施,保障受试者安全。密切注意药物不良反应,按照规定进行报告和处理。出现大范围、非预期的药物不良反应,或确证临床试验药物有严重质量问题,SFDA或省级药品监督管理部门,可以责令暂停或终止临床研究。生物等效性试验与已生产对照药品的生物利用度比较试验。要求:盲法或开放试验;病例:18-24例新药的申报与审批(一)新药申报与审批程序新药注册申报与审批,分为临床研究申报审批和生产上市申报审批。省级药品监督管理部门负责初审指定的药检所负责样品检验和申报的药品标准复核SFDA负责对新药进行技术审批和所有资料的全面审评1、新药临床研究的审批程序2、新药生产的审批程序(二)新药审批有关规定1.对下列新药申请可实行快速审批(1)新的中药材及其制剂,中药或者天然药物中提取的有效成分及其制剂(2)未在国内外获准上市的化学原料药及其制剂、生物制品(3)抗艾滋病病毒及用于诊断、预防艾滋病的新药,治疗恶性肿瘤、罕见病等的新药。(4)治疗尚无有效治疗手段的疾病的新药。(5)突发事件应急所必需的药品。2.对报送材料的要求•申请新药注册所报送的资料应当完整、规范、数据必须真实、可靠;•引用文献资料应当注明著作名称、刊物名称及卷、期、页等;•未公开发表的文献资料应当提供资料所有者许可使用的证明文件。•外文资料应当按照要求提供中文译本。3.联合研制的新药申报•多个单位联合研制的新药,可以由其中的一个单位申请注册,其他的单位不得重复申请。需要联合申请注册的,应当共同署名作为该新药的申请人。•除加快审批创新药物外,新药申请批准后每个品种只能由一个单位生产,同一品种的不同规格不得分由不同单位生产。新药的技术转让新药技术转让,是指新药证书的持有者,将新药生产技术转让给药品生产企业,并由该药品生产企业申请生产该新药的行为。新药技术的转让方应当是新药证书的持有者。转让方已取得药品批准文号的,申请新药技术转让时,应当同时提出注销其药
 三七文档所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
三七文档所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
 扫描二维码
扫描二维码







 雷na
雷na
本文标题:新药审批
链接地址:https://www.777doc.com/doc-364540 .html