您好,欢迎访问三七文档
当前位置:首页 > 商业/管理/HR > 商业合同/协议 > 药品出厂、上市放行管理规程
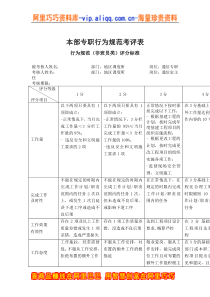
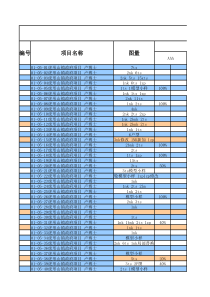
XXXXXXXXXX有限公司质量保证管理制度标题药品出厂、上市放行管理规程共4页第1页文件号起草人起草日期部门审阅日期QA审阅日期批准日期生效日期颁发部门分发部门变更记录文件修订号变更版本变更时间变更原因1目的:依据2019年新版《药品管理法》第三十二条、第四十七条之规定,建立药品出厂、上市放行管理规程,确保出厂、上市后的药品质量。2范围:本规程适用于本公司生产的所有药品放行的管理。3责任:质量受权人、质保部、生产部、生车间相关人员。4内容:4.1药品出厂、上市审核放行要求:4.1.1药品出厂、上市放行前必须由生产部进行生产审核,由质保部质量审核人员对有关记录进行审核并签字,其审核内容包括:配重、称重过程中的复核情况;各生产工序生产检查记录;清场记录;中间产品质量检验结果;偏差处理;成品检验结果等项。4.1.2药品审核符合要求后,由质量受权人批准后方可放行。4.2生产部审核的内容及标准4.2.1批生产指令中配方与工艺规程是否相符,生产指令单上应有指令下达人、质量审核人、指令接收人、QA和批准人的签字。4.2.2所使用的物料有无检验报告单。4.2.3领料单与处方应相符,物料领用数量应与领料单相符,投料量、投料次序均应符合现行工艺规程。药品出厂、上市放行管理规程第2页共4页4.2.4生产过程应符合现行工艺规程,并执行批准的标准操作规程。4.2.5清场记录填写是否完整,并附有清场标识。4.2.6生产设备状态标识是否符合规定要求。4.2.7批生产记录填写正确、完整无误,各项内容及数据均符合标准规定;4.2.8物料平衡计算公式是否正确,各工序物料平衡结果是否符合规定限度。4.2.9中间产品检验结果是否符合质量检验标准要求;4.2.10包装时所用说明书、合格证应正确;打印的生产批号、生产日期及复验期(有效期)应正确。4.2.11包装记录应齐全、书写无误、数据完整、准确,并有操作人、复核人和QA签名。4.2.12人员卫生和生产环境监测是否符合规定。4.2.13生产过程监控项目齐全,结果是否符合规定。监控的记录是否齐全,并有监控人签名。4.2.14如发生偏差、变更和检验结果超标时,按程序处理并有充分说明。4.3QC、QA4.3.1QC对批检验记录的审核4.3.1.1原始记录填写是否规范,原始数据是否准确、真实,数据的处理是否符合相关规程。4.3.1.2检验的原始图谱是否齐全,是否有检验人、复核人签名及日期,原始数据图谱与记录数据应一致。4.3.2QC对检验过程的复核4.3.2.1检验仪器和设备是否经过校验,是否在效期内。4.3.2.2玻璃计量器具是否经标定,是否在效期内。药品出厂、上市放行管理规程第3页共4页4.3.2.3试液、滴定液和对照品是否在效期内,是否有使用记录。4.3.2.4检验方法是否经过确认。4.3.2.5车间环境监控数据是否合格。4.3.2.6检验报告单检验项目是否完整、结果是否符合标准。4.3.2.7药品生产所使用物料的检验报告单项目是否完整、结果是否符合标准。4.3.2.8若有检验偏差,检验偏差是否按照相关规程进行处理。4.3.3QA对生产、检验记录审核及评价4.3.3.1QA应对生产部和QC审核后的内容进行再次质量审核和评价。4.3.3.1.1以QA对生产各环节生产记录审核结果为评价依据,若生产记录缺失、数据不完整等均属生产过程不合格。4.3.3.1.2以QA对批检验记录、检验结论(依据相关检验标准规定)审核结果为依据,若检验记录缺失、数据不完整等均属检验不合格。4.4药品质量评价标准4.3.3.2.1合格药品:经审核生产和检验均符合标准要求的药品。4.3.3.2..2不合格药品:经审核生产和检验均不符合标准要求或其某项不合格的药品即可评价为不合格药品。4.4药品出厂、上市放行的批准4.4.1经质量审核和评价确认为合格药品后,由质保部QA盖印有“药品经质量受权人批准准予放行”字样的放行章,经质量受权人审核签名确认后,药品方可放行。4.4.2质量受权人将对所签署的《药品出厂、上市放行审核单》(ZL-086)所述内容的真实性、完整性负责。4.4.3质量受权人需对其批准出厂、上市的药品质量安全负责。药品出厂、上市放行管理规程第4页共4页4.4.4质量受权人属最终产品放行人,不允许转授权。4.4.5经质量审核和评价确认为不合格药品后,签发不合格品报告单,经质量受权人签名确认后,按不合格品管理程序执行。
 三七文档所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
三七文档所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
 扫描二维码
扫描二维码








 as051204011
as051204011
本文标题:药品出厂、上市放行管理规程
链接地址:https://www.777doc.com/doc-7305452 .html