当前位置:首页 > 行业资料 > 国内外标准规范 > GB 14758-2010 食品添加剂 咖啡因
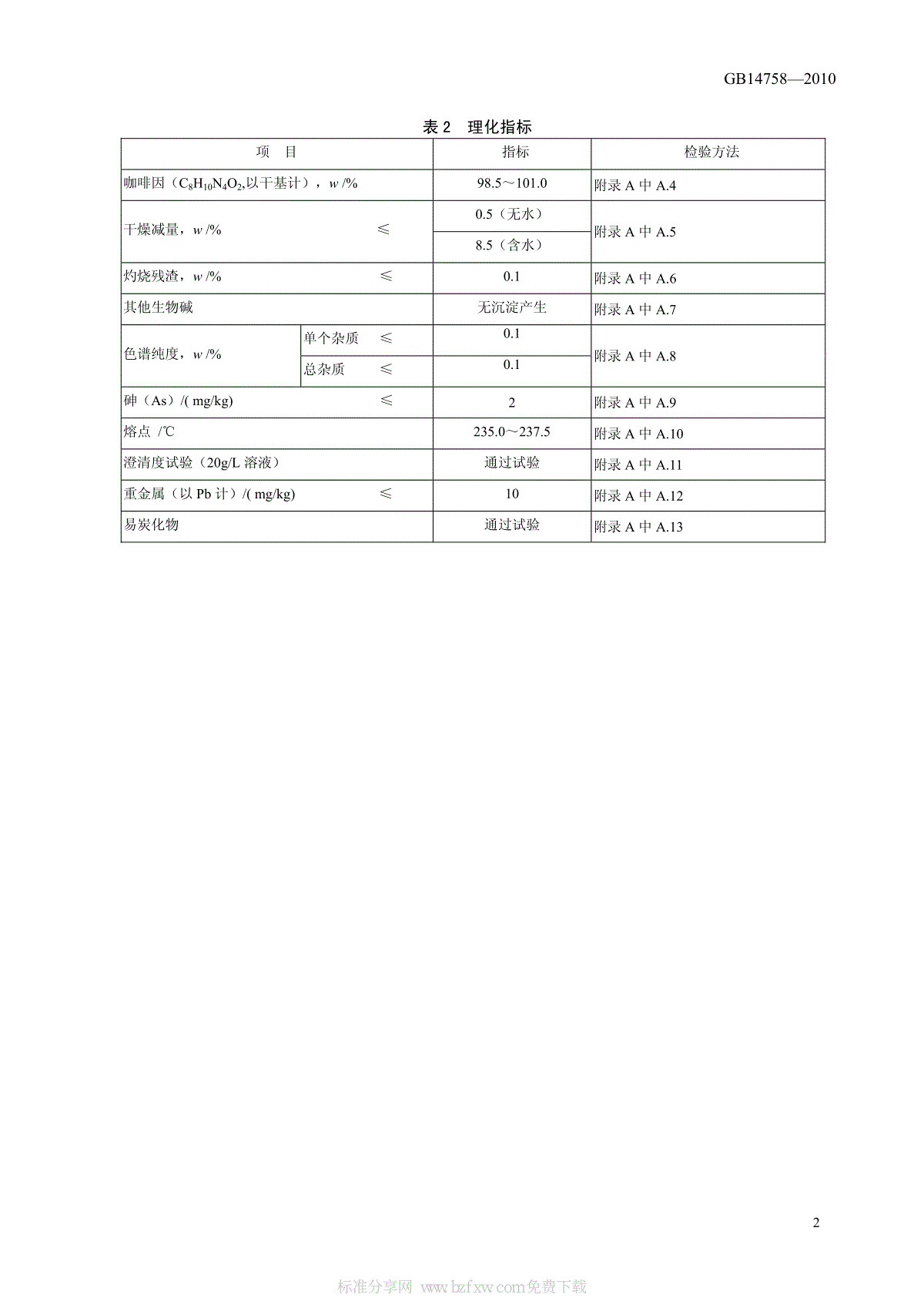
中华人民共和国国家标准GB14758—2010食品安全国家标准食品添加剂咖啡因2010-12-21发布2011-02-21实施中华人民共和国卫生部发布GB14758—2010I前言本标准代替GB14758—1993《食品添加剂咖啡因》。本标准与GB14758—1993相比,主要变化如下:——外观描述由白色结晶粉末改为白色或极微黄绿色结晶粉末;——咖啡因含量的允许差由相对差0.3%改为绝对差0.2%;——增加了红外光谱鉴别;——增加了澄清度项目;——砷指标由0.0003%修改为2mg/kg;——增加了色谱纯度的要求。本标准的附录A和附录B为规范性附录,附录C为资料性附录。本标准所代替标准的历次版本发布情况为:——GB14758-1993GB14758—20101食品安全国家标准食品添加剂咖啡因1范围本标准适用于以氯乙酸或氰乙酸为原料化学合成法及以茶叶为原料提取法制得的食品添加剂咖啡因。2规范性引用文件本标准中引用的文件对于本标准的应用是必不可少的。凡是注日期的引用文件,仅所注日期的版本适用于本标准。凡是不注日期的引用文件,其昀新版本(包括所有的修改单)适用于本标准。3化学名称、分子式、结构式和相对分子质量3.1化学名称1,3,7-三甲基黄嘌呤3.2分子式C8H10N4O2或C8H10N4O2·H2O3.3结构式3.4相对分子质量无水咖啡因:194.19(按2007年国际相对原子质量)一水咖啡因:212.21(按2007年国际相对原子质量)4技术要求4.1感官要求:应符合表1的规定。表1感官要求项目要求检验方法色泽白色或带极微黄绿色且有丝光取适量样品置于清洁、干燥的烧杯中,在自然光线下,观察色泽和组织状态,嗅其气味。用温开水漱口后,品尝滋味。滋味、气味味苦,无臭组织状态针状结晶或结晶性粉末或颗粒。具有风化性4.2理化指标:应符合表2的规定。CH3NNNNCH3CH3OOGB14758—20102表2理化指标项目指标检验方法咖啡因(C8H10N4O2,以干基计),w/%98.5~101.0附录A中A.4干燥减量,w/%≤0.5(无水)附录A中A.58.5(含水)灼烧残渣,w/%≤0.1附录A中A.6其他生物碱无沉淀产生附录A中A.7色谱纯度,w/%单个杂质≤0.1附录A中A.8总杂质≤0.1砷(As)/(mg/kg)≤2附录A中A.9熔点/℃235.0~237.5附录A中A.10澄清度试验(20g/L溶液)通过试验附录A中A.11重金属(以Pb计)/(mg/kg)≤10附录A中A.12易炭化物通过试验附录A中A.13GB14758—20103附录A(规范性附录)检验方法A.1安全提示本标准试验方法中使用的部分试剂具有毒性或腐蚀性,按相关规定操作,使用时需小心谨慎。若溅到皮肤上应立即用水冲洗,严重者应立即治疗。在使用挥发性酸时,要在通风橱中进行。A.2一般规定本标准所用试剂除非另有说明,在分析中仅使用确认为分析纯的试剂和GB/T6682—2008中规定的三级水。试验方法中所用标准滴定溶液、杂质测定用标准溶液、制剂及制品,在没有注明其他要求时,均按GB/T601、GB/T602、GB/T603的规定制备。A.3鉴别试验A.3.1试剂和材料A.3.1.1盐酸。A.3.1.2氯酸钾。A.3.1.3氨溶液:10→100。A.3.1.4盐酸溶液:10→100。A.3.1.5氢氧化钠溶液:40g/L。A.3.1.6碘溶液:0.1mol/L。A.3.2分析步骤A.3.2.1紫脲酸呈色试验取约10.0mg实验室样品,加1mL盐酸溶液与0.1g氯酸钾,置水浴上蒸干,残渣遇氨气即显紫色,再加氢氧化钠溶液数滴,紫色即消失。A.3.2.2咖啡因与碘试液及盐酸的沉淀反应取5mL实验室样品的饱和水溶液,加5滴碘溶液,不发生沉淀;再加3滴盐酸溶液,即出现红棕色的沉淀,能在稍过量的氢氧化钠溶液中溶解。A.3.2.3红外光谱鉴别采用溴化钾压片法,按照GB/T6040进行试验,实验室样品的红外光谱应与对照图谱(见附录B)一致。A.4咖啡因的测定A.4.1试剂和材料A.4.1.1乙酸酐。A.4.1.2苯。A.4.1.3高氯酸标准滴定溶液:c(HClO4)=0.1mol/L。GB14758—20104A.4.2仪器和设备电位滴定仪。A.4.3分析步骤取约0.4g实验室样品,精确至0.0002g,加40mL乙酸酐,加热溶解,冷却,加80mL苯,用高氯酸标准滴定液滴定,用电位法指示终点,并将滴定的结果用空白试验校正。A.4.4结果计算咖啡因(以C8H10N4O2计)的质量分数w1,数值以%表示,按公式(A.1)计算:012100(11000VVcMwmw−××=××−×())%………………………………(A.1)式中:V——实验室样品消耗高氯酸标准溶液的体积,单位为毫升(mL);V0——空白试验消耗高氯酸标准溶液的体积,单位为毫升(mL);c——高氯酸滴定液的摩尔浓度,单位为摩尔每升(mol/L);m——实验室样品的质量的数值,单位为克(g);w2——干燥减量的数值,%;M——咖啡因的摩尔质量的数值,单位为克每摩尔(g/mol)[M(C8H10N4O2)=194.2]取平行测定结果的算术平均值为测定结果,两次平行测定的绝对差值不大于0.2%。A.5干燥减量的测定A.5.1分析步骤取1.0g实验室样品,精确至0.0002g,置于已在80℃干燥至恒重的称量瓶内,精密称量,置于80℃烘箱中干燥至恒重。A.5.2结果计算样品干燥减量的质量分数以2w计,数值以%表示,按公式(A.2)计算:12213100mmwmm−=×−%……………………………………(A.2)式中:m1——干燥前实验室样品和称量瓶总质量的数值,单位为克(g);m2——干燥后实验室样品和称量瓶总质量的数值,单位为克(g);m3——称量瓶质量的数值,单位为克(g)。取平行测定结果的算术平均值为测定结果,两次平行测定结果的绝对差值不大于0.05%。A.6灼烧残渣的测定A.6.1试剂和材料硫酸。A.6.2分析步骤取1.0g实验室样品,精确至0.001g,置于已在750℃±50℃烧至恒重的瓷坩埚中,用小火缓缓加热至完全炭化。冷却至室温后,加硫酸0.5mL~1mL使湿润,低温加热至硫酸蒸气除尽后,移入高温炉中,在750℃±50℃高温炉中灼烧至恒重。GB14758—20105A.6.3结果计算灼烧残渣的质量分数以3w计,数值以%表示,按公式(A.3)计算:453100mmwm−=×%…………………………………(A.3)式中:m4——坩埚和残渣总质量的数值,单位为克(g);m5——坩埚质量的数值,单位为克(g);m——样品质量的数值,单位为克(g)。取平行测定结果的算术平均值为测定结果,两次平行测定结果的绝对差值不大于0.02%。A.7其他生物碱的测定A.7.1试剂和材料碘化汞钾试液:取1.36g二氯化汞,加60mL水使溶解,另取5g碘化钾,加10mL水使溶解,将两液混合,混合后加水稀释至100mL。A.7.2分析步骤称取1g实验室样品,加50mL水,取10mL该溶液加3滴碘化汞钾试液,不得产生沉淀。A.8色谱纯度的测定A.8.1试剂和材料A.8.1.1无水乙酸钠。A.8.1.2乙腈:色谱纯。A.8.1.3四氢呋喃:色谱纯。A.8.1.4冰乙酸。A.8.1.5茶碱对照品。A.8.1.6咖啡因对照品。A.8.2仪器和设备高效液相色谱仪。A.8.3色谱分析条件推荐的色谱柱及典型色谱操作条件见表A.1,咖啡因系统适用性试验高效液相色谱图参见图C.1,各组分的相对保留时间参见表C.1。其他能达到同等分离程度的色谱柱和色谱条件均可使用。φ表A.1色谱柱和典型色谱操作条件色谱柱十八烷基硅烷键合硅胶色谱柱(填料粒径5µm,φ4.6mm×150mm不锈钢柱)流动相称取约1.64g无水乙酸钠,精密称定,加水溶解并稀释至2000mL,混匀。取1910mL,加乙腈50mL,四氢呋喃40mL,混合后用冰乙酸调pH=4.5,混匀,过滤,脱气流速约1mL/min检测器检测波长275nmA.8.4分析步骤A.8.4.1对照品溶液:称取约2mg茶碱对照品,精确至0.0001g,置100mL容量瓶中,加约50mL流动相振摇,并用超声波溶解,用流动相稀释到刻度,混匀。GB14758—20106A.8.4.2系统适用性试液的制备:称取约5.0mg咖啡因对照品,精确至0.0001g,置25mL容量瓶中,加5.0mL对照品溶液和10mL流动相,振摇,超声溶解,再用流动相稀释到刻度,混匀。A.8.4.3实验室样品液的制备:称取约10mg咖啡因实验室样品,精确至0.0001g,置50mL容量瓶中,加10mL流动相,振摇,超声溶解,再用流动相稀释到刻度,混匀。A.8.4.4仪器准备:按照高效液相色谱仪操作规程准备仪器,设定流速为1mL/min,检测波长为275nm,用流动相平衡仪器后,开始进样操作。A.8.4.5系统适应性试验:取10µL系统适用性试液进样,记录色谱图,茶碱和咖啡因的相对保留时间依次约为0.69,1.0;两峰间的分离度不得小于6.0;每个峰的拖尾因子T≤2.0。A.8.4.6测定:取流动相和样品溶液各10µL进样,记录色谱图至咖啡因主峰保留时间的两倍。按面积归一化法计算各杂质含量。A.8.5结果计算(面积归一化法)杂质含量的质量分数以4w计,数值以%表示,按公式(A.4)计算:4×100AwA=∑杂%………………………………(A.4)式中:A杂——杂质峰的峰面积(除溶剂峰及主峰外);∑A——所有峰的面积之和(溶剂峰除外)。A.9砷的测定取1.0g±0.01g实验室样品,加20mL水,加热溶解,冷却至室温后转移至100mL锥形瓶中,加5mL盐酸、5mL碘化钾试液,5滴酸性氯化亚锡溶液,按照GB/T5009.76—2003砷斑法的规定进行。A.10熔点的测定按《中华人民共和国药典》2005年版二部附录VIC熔点测定法第一法进行。方法如下:取实验室样品适量,研成细粉,在80℃烘箱中干燥4h后,分取适量,置熔点测定用毛细管(简称毛细管,由中性硬质玻璃管制成,长9cm以上,内径0.9mm~1.1mm,壁厚0.10mm~0.15mm,一端熔封;当所用温度计浸入传温液在6cm以上时,管长应适当增加,使露出液面3cm以上)中,轻击管壁或借助长短适宜的洁净玻璃管,垂直放在表面皿或其他适宜的硬质物体上,将毛细管自上口放入使自由落下,反复数次,使粉末紧密集结在毛细管的熔封端。装入实验室样品的高度为3mm。另将温度计(分浸型,具有0.5℃刻度,经熔点测定用对照品校正)放入盛装传温液(硅油或液状石蜡)的容器中,使温度计汞球部的底端与容器的底部距离2.5cm以上(用内加热的容器,温度计汞球与加热器上表面距离2.5cm以上);加入传温液以使传温液受热后的液面适在温度计的分浸线处。将传温液加热,待温度上升至较规定的熔点低限约低10℃时,将装有实验室样品的毛细管浸入传温液,贴附在温度计上(可用橡皮圈或毛细管固定),位置须使毛细管的内容物适在温度计汞球中部;继续加热,调节升温速率为每分钟上升1.0℃~1.5℃,加热时须不断搅拌使传温液温度保持均匀,记录实验室样品在初熔至全熔时的温度,重复测定3次,取其平均值。A.11澄清度的测定A.11.1试剂和材料A.11.1.1乌洛托品溶液:10g/L。GB14758—20107A.11.1.2浊度标准贮备液的制备:称取于105℃干燥至恒重的1.00g硫酸肼,置100mL量瓶中,加水适量使溶解,必要时可在40℃的水浴中温热溶解,并用水稀释至刻度,摇匀,放置4h~6h;取此溶液与等容量的乌洛托品溶液(100g/L)混合,摇匀,于25℃避光静置24h,即得。本液置冷处避光保存,可在两个月内使用,用前摇匀。A.11.1.3浊度标准原液的制备:取15.0mL浊度标准贮备液,置1000mL量瓶中,加水稀释至刻度,摇匀,取适量,置1cm吸收池中,照紫外-可见分光光度法(《中华人民共和国药典》2005年版附录IVA),在550nm的波长处测定,其吸光度应在0.1




 三七文档所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
三七文档所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
 扫描二维码
扫描二维码


![107[高考试题]机械振动 机械波](/doc-111726.png)







 ●smile、兰
●smile、兰
本文标题:GB 14758-2010 食品添加剂 咖啡因
链接地址:https://www.777doc.com/doc-9400403 .html